7.1. Методы предварительного анализа
7.1.1. Понятие об аналитическом скрининге в химико-токсикологическом анализе
7.1.2. ТСХ-скрининг в нормально-фазовом варианте
7.1.3. ТСХ-скрининг в варианте «Toxi-Lab АВ»
7.1.4. ТСХ-скрининг в обращенно-фазовом варианте (ОФТСХ)
7.1.5. Внутригрупповой ТСХ-скрининг в частных системах растворителей
7.1.6. Газожидкостная хроматография
7.1.6.1. ГЖХ-скрининг в анализе лекарственных и наркотических веществ в извлечениях из мочи
7.1.6.2. ГЖХ-скрининг в анализе «летучих» ядов
7.1.7. Иммунохимические методы скрининга лекарственных и наркотических веществ
7.1.8. Аналитический скрининг с помощью химических реакций
7.2. Методы подтверждающего анализа
7.2.1. Метод высокоэффективной жидкостной хроматографии (ВЭЖХ)
7.2.2. Методы ПК- и УФ-спектроскопии
7.2.3. Хроматомасс-спектрометрия
7.2.4. Люминесцентный метод анализа
7.2.5. Микрокристаллоскопический метод
7.2.6. Фармакологические (физиологические) пробы
7.2.7. Фармакогностический анализ
Обнаружение ядовитых, сильнодействующих и наркотических веществ после их изолирования из биологических объектов является следующим этапом химикотоксикологического исследования.
Несмотря на большое число реакций и методов качественного анализа, используемых в аналитической и фармацевтической химии, требуются некоторые особые подходы для их использования в токсикологической химии. Это объясняется особенностями объектов исследования, возможными примесями и требованиями к достоверности результатов.
Химик-токсиколог для обнаружения в объекте какой-либо группы веществ или определенного вещества обязан использовать только те реакции и методы, которые апробированы на биологических объектах, рекомендованы для целей химико-токсикологического анализа с учетом всех факторов, влияющих на получаемые результаты. Только в этом случае полученные данные могут быть объективными и застрахованными от возможных ошибок, среди которых могут быть ошибки, связанные с получением ложноположительных или ложноотрицательных результатов.
Ложноположительный результат — это заключение о наличии токсических, экзогенных веществ при анализе по определенной методике при фактическом их отсутствии. Это свидетельствует об использовании высокочувствительной, недостаточно специфичной методики и возможном отсутствии ее апробации на биологических объектах, особенно находящихся в стадии гнилостного разложения.
Если какое-либо вещество или его метаболит присутствуют в объекте в концентрации ниже предела их обнаружения, то возможно получение ложноотрицательных результатов. Причиной этого чаще всего выступает фактор времени, т.е. интервал времени между контактом ядовитого вещества с организмом человека или животного и временем взятия и направления объекта на анализ. В этом случае даже правильно выбранная методика с высокой чувствительностью может дать отрицательный результат.
Для обнаружения токсических соединений в биологических объектах в настоящее время применяются различные химические реакции, физические, физико-химические, биологические (фармакологические) и другие методы. Среди них выделяют методы и реакции предварительного анализа и методы и реакции подтверждающего исследования.
Использование предварительного анализа или предварительных химических реакций преследует цель обнаружить или исключить из анализа группу веществ или какое-либо индивидуальное вещество. Таким реакциям или методам придают «судебно-химическое значение при отрицательном результате». Это означает, что если при использовании данного метода или реакции вещество или группа веществ не обнаружены, дальнейший анализ на эти соединения не проводят и в заключении указывают, что данное вещество или группа веществ в исследуемом объекте не найдены.
Предварительные реакции и методы должны быть чувствительными, но необязательно специфичными. Среди них — скрининговые методы (ТСХ, Г’ЖХ — скрининг, им- мунохимические методы) и реакции, которые оцениваются как имеющие значение при отрицательном результате: групповые реакции осаждения, хромогенные реакции и др. Эти способы и реакции должны быть обязательно подтверждены химическими и физикохимическими методами.
Предварительные реакции и методы, как правило, не позволяют определить индивидуальные вещества. Это требует проведения дополнительных исследований. В токсикологической химии такие исследования называют подтверждающим анализом, подтверждающими (частными) реакциями или частным внутригрупповым скринингом. Среди них методы ВЭЖХ, ГЖХ, ТСХ, УФ- и ИК-спектрометрия, спектрофотометрия в УФ области спектра, масс-спектрометрия, хроматомасс-спектрометрия, люминесцентный анализ, аналитические реакции разных типов (кислотно-основные, окислительно- восстановительные, комплексообразования), микрокристаллоскопический метод, фармакогностический анализ, фармакологические пробы, а также процессы осаждения, экстракции и др.
Подтверждающие методы по чувствительности обычно выше или равны предварительным методам исследования. Это позволяет снизить возможность получения ложноотрицательных результатов. По специфичности подтверждающие методы обычно превосходят предварительные, что снижает количество ложноположительных результатов.
При проведении химико-токсикологического анализа с использованием химического метода необходимо учитывать следующие обстоятельства.
Любое чужеродное вещество в организме частично или полностью подвергается метаболизму, и поэтому его можно не обнаружить известными реакциями.
В процессе изолирования ядовитые вещества извлекаются не полностью и могут быть частично потеряны, поэтому используемые реакции должны отличаться высокой чувствительно стью.
Объекты, присылаемые на анализ, могут находиться в состоянии гнилостного разложения. Продукты разложения (птомаины) обычно извлекаются из объекта вместе с исследуемым веществом и могут искажать результаты анализа. Поэтому при анализе таких объектов использование химических реакций возможно после тщательной очистки извлечений.
При комбинированных отравлениях (например, этиловым спиртом и его суррогатами) применение характерных реакций затруднено присутствием близких по химическим свойствам соединений. В этих случаях предусмотрено строгое соблюдение условий воспроизведения реакций (окисления, температурного режима и т.д.), чтобы предотвратить обнаружение одних соединений (более ядовитых) вместо других. Например, при несоблюдении условий проведения реакции окисления можно переоткрыть метиловый спирт за счет этилового спирта.
Каждая используемая химическая реакция должна быть рекомендована в определенной модификации применительно к исследуемому объекту. Это особенно важно в тех случаях, когда проводят исследование объекта на «металлические» яды, поскольку многие из них являются микроэлементами и могут быть обнаружены в извлечениях. В таблице 5 даны пределы содержания токсикологически важных элементов в печени и почках — объектах, которые рекомендуются для анализа при подозрении на отравление «металлическими» ядами и содержат наибольшее количество микроэлементов.
В связи с варьированием содержания элементов в тканях, для количественного определения металлов в минерализате для каждого катиона предложены не менее двух методов, которые позволяют определять их в широких пределах концентраций.
По схеме дробного метода анализа предусматривается разбавление минерализата до концентраций, когда естественно содержащийся микроэлемент не может быть обнаружен, а концентрация попавшего в организм элемента останется достаточной для его обнаружения. При проведении реакций к минерализату добавляют определенный объем воды очищенной, а затем вносят соответствующие реактивы. Приведенные в таблице 6 разведения позволяют исключить обнаружение микроэлементов и одновременно снизить кислотность минерализата.
Таблица 5. Пределы содержажания некоторых элементов в тканях организма человека (по данным А.Н. Крыловой)
|
Название элемента |
Границы естественного содержания, мг на 100 г органа |
|
|
Печень |
Почки | |
|
Ртуть |
0-0,01 |
0-0,04 |
|
Свинец |
0,13 |
0,027 |
|
Марганец |
0,13-0,40 |
0,06-0,28 |
|
Хром |
0,001-0,013 |
0,028-0,029 |
|
Серебро |
0,005 |
— |
|
Медь |
0,56-1,12 |
0,26-0,40 |
|
Мышьяк |
0,01-0,07 |
0,01-0,08 |
|
Кадмий |
0,64-6,68 |
1,32-8,48 |
|
Цинк |
2,73-6,71 |
1,76-6,16 |
|
Барий |
Незначительные количества |
|
|
Висмут |
Следы |
|
|
Сурьма |
Не обнаружена |
|
|
Таллий |
Не обнаружен |
|
|
Железо |
До 200 |
|
|
Кальций |
До 100 |
|
|
Натрий |
Значительные количества |
|
|
Калий |
Значительные количества |
|
|
Фосфаты |
Значительные количества |
|
|
Хлориды |
Небольшие количества |
|
Таблица 6. Разбавление минерализата при анализе его на «металлические» яды
|
Название элемента |
Объем минерализата, |
Объем воды |
Граница обнаружения, мг/100 г |
|
Марганец |
1,0 |
1,0 |
0,04 |
|
Хром |
6,0 |
1.0 |
0,10 |
|
Серебро |
5,0 |
— |
0,05 |
|
Медь |
10,0 |
— |
0,40 |
|
Кадмий |
10,0 |
— |
2,00 |
|
Сурьма |
1,0 |
10 |
0.10 |
|
Висмут |
10,0 |
10 |
0,10 |
|
Цинк |
0,5 |
10 |
5,00 |
|
Мышьяк |
2,0 |
20 |
0,01 |
При анализе минерализата на «металлические» яды применяется избирательная экстракция в виде диэтилдитиокарбаматов с целью выделения меди, висмута, цинка и кадмия в органическую фазу, свободную от мешающих ионов. Специфичность экстракционных методик выделения этих катионов достигнута за счет использования правила ряда диэтилдитиокарбаматов металлов:
Hg2+, Аg+, Cu2+, Ni2+, Со2+, Bi3+, Sb3+, Cd2+, Pb2+, Zn2+, Mn2+
Согласно этому ряду каждый предыдущий металл вытесняет последующий из его соли с диэтилдитиокарбаминовой кислотой.
Таблица 7. Маскировка некоторых мешающих ионов в химических реакциях на исследуемые металлы
|
Используемые ионы |
Комплексное соединение |
|
Цианиды |
[Men+(CNm)](mn) Со2+, Zn2+, Fe3+, Сd2+, Fe2+, Hg2+, Ag+ |
|
Фториды |
[FeF6]3- |
|
Фосфаты |
[Fe(PO4)2]3- |
|
Тиосульфаты |
[Men+(S2O3)m]-(2m-n) Ag+, Pb2+, Fe2+, Bi2+, Fe3+, Sb3+, Cd2+, Hg2+ |
|
Тиомочевина |
[Men+(S=C(NH2)2)m]n+ Bi3+, Fe3+, Sb3+, Cd2+, Hg2+, Ag+ и др. |
Селективная экстракция с дитизоном используется для обнаружения ионов свинца, серебра, таллия, цинка, которые при определенном значении pH среды образуют окрашенные дитизонаты.
Для исключения влияния других ионов на результаты реакций при анализе минерализата используют прием маскировки мешающих ионов (табл. 7). С этой целью используют цианиды, фториды, фосфаты, тиосульфаты, тиомочевину, гидроксиламин, аскорбиновую кислоту, комплексон III (трилон Б) и др. Мешающие ионы образуют с этими веществами комплексные соединения и не влияют на результаты реакций, проводимых на исследуемые элементы.
Абсолютно специфичных реакций очень мало. По правилам проведения химикотоксикологического анализа для заключения об обнаружении в извлечении из объекта ядовитого соединения необходимо провести не менее 3-4 характерных для данного вещества реакций. Например, чтобы сделать вывод об обнаружении в дистилляте этилового спирта, необходимо, чтобы три реакции дали положительный результат: образование йодоформа, окисление спирта до уксусного альдегида и образование эфира с уксусной кислотой (этилацетата).
Результаты некоторых химических реакций оцениваются как «реакции Corpus delicti». Это реакции, продукты взаимодействия которых можно представить вместе с заключением об отравлении как неоспоримое доказательство обнаружения в объекте данного вещества. К числу таких реакций относятся: реакция образования берлинской лазури, результат обнаружения мышьяка в аппарате Марша, реакция образования «серебряного зеркала» и др. Полученный синий осадок берлинской лазури запаивают в небольшую пробирку или ампулу и представляют судебно-следственным органам для обоснованности заключения об обнаружении в объекте синильной кислоты. Восстановительная трубка аппарата Марша и микрофотографии налета на ней, имеющего характерную форму кристаллов в виде октаэдров, убедительное доказательство обнаружения мышьяка в исследуемом объекте.
Для увеличения избирательности некоторых реакций и повышения их чувствительности иногда рекомендуется предварительное выделение ядовитого вещества из исследуемого раствора с использованием экстракции и реэкстракции. Например, чтобы отделить изоамиловый спирт от других «летучих» ядов и воды, его из дистиллята извлекают диэтиловым эфиром, эфир испаряют и в остатке обнаруживают изоамиловый спирт соответствующими реакциями. Фенол выделяют из дистиллята, подщелоченного гидрокарбонатом натрия, также путем экстракции диэтиловым эфиром, что позволяет повысить избирательность и чувствительность реакций на него.
При химическом анализе на любое чужеродное соединение эксперт должен руководствоваться специально изданными и утвержденными методиками и соответствующими методическими указаниями.
Комплексное использование различных методов предварительного и подтверждающего анализов позволяет надежно диагностировать причину отравления или болезненного состояния организма.
Обнаружение ядовитых веществ и их метаболитов в процессе предварительного и подтверждающего химико-токсикологического анализа не дает однозначного ответа о количестве яда, попавшего в организм, времени его приема, фазах распределения. Результаты количественного определения найденного токсического соединения позволяют выбрать способ детоксикации и лечения пострадавшего при остром и хроническом отравлении.
Выбор метода количественного определения ядовитого вещества в извлечении из объекта зависит:
— от предполагаемой концентрации токсического вещества по результатам предварительного и основного (подтверждающего) исследования;
— от степени «свежести» объекта и наличия в извлечении продуктов разложения белковых молекул;
— от времени, прошедшего от момента попадания яда в организм и до начала анализа;
— от степени «чистоты» извлечения и наличия фона эндогенных соединений;
— от способности токсического вещества и его метаболитов накапливаться в определенных органах и тканях;
— от последствий отравления. В случае смертельного исхода чувствительность методик должна быть 10-3 — 10-7 г/мл. При наркотическом опьянении, затяжном отравлении или после проведенных детоксикационных мероприятий — 10-8 — 10-13 г/мл.
7.1. Методы предварительного анализа
7.1.1. Понятие об аналитическом скрининге в химикотоксикологическом анализе
Многообразие лекарственных, наркотических, ядовитых веществ, которые могут быть причиной отравлений, требует проведения предварительного отсеивающего исследования. В токсикологической химии такие исследования принято называть аналитическим скринингом. Скрининг (screening) в переводе с английского означает «просеивание», «отбор», «сортировка».
Аналитический скрининг — это система методических приемов, позволяющих в ходе исследовательских операций исключить («отсеять») или определить группы веществ (индивидуальные соединения) на этапе предварительного исследования.
Это позволяет построить дальнейший анализ в нужном направлении. Аналитический скрининг является эффективным при его соответствии определенным требованиям:
— специфичность (чаще групповая);
— высокая чувствительность;
— экспрессность;
— точность и воспроизводимость;
— возможность сочетания с другими методами анализа.
Скрининг используется при анализе многокомпонентных смесей, а в случае ненаправленного анализа — при исследовании извлечения из биологического объекта на неизвестное токсическое вещество.
В настоящее время понятие скрининга в токсикологической химии значительно шире.
Скрининг — это поэтапное движение к выявлению индивидуального вещества путем последовательного исключения групп ядовитых веществ, а затем отсеивания веществ в обнаруженной группе до выявления конкретного соединения.
Из современных скрининговых методов в практике химико-токсикологического анализа нашла широкое применение хроматография в тонких слоях сорбента (ТСХ) в нормально-фазовом и обращенно-фазовом вариантах. Этот метод доступен, прост в выполнении, отличается высокой чувствительностью, эффективностью, экспрессностью и достаточной специфичностью (избирательностью).
ГЖХ-скрининг используется, в основном, при анализе летучих, лекарственных и наркотических веществ.
Не потерял своего значения аналитический скрининг с использованием различных химических реакций.
Имеются разработки по использованию в качестве скрининговых методов высокоэффективной жидкостной хроматографии (ВЭЖХ), абсорбционной спектроскопии, им- мунохимических методов, спектроскопии ядерного магнитного резонанса.
При использовании этих методов необходима тщательная очистка извлечений от эндогенных соединений, способных исказить результаты анализа и засорить колонки хроматографов.
7.1.2. ТСХ-скрининг в нормально-фазовом варианте
Метод используется в химико-токсикологическом анализе при исследовании веществ, изолируемых из объекта экстракцией и сорбцией (лекарственные, наркотические вещества, пестициды).
Неподвижной фазой в ТСХ служит тонкий слой сорбента (0,1-0,5 мм), содержащий небольшое количество воды и нанесенный на пластинку (из стекла, фольги или полимера).
Сорбентом чаще всего является силикагель или оксид алюминия, которые закрепляются на пластинке добавлением связующего компонента — гипса, крахмала и др.
В качестве подвижной фазы (элюента, системы растворителей) предложены индивидуальные растворители или их смеси в определенных соотношениях. При движении элюента за счет капиллярных сил вверх по пластинке происходит разделение смеси веществ. Эффективность разделения зависит от сродства вещества к сорбенту и определяется коэффициентом распределения его между обеими фазами — подвижной и неподвижной.
Чтобы обнаружить вещество на пластинке, используют различные способы детектирования:
— визуальный, если само вещество окрашено;
— облучение пластинки УФ-лучами, при этом вещество может флуоресцировать или давать темные пятна;
— обработка пластинки соответствующими реагентами-проявителями, которые способны образовать с веществом окрашенные соединения.
Для идентификации вещества используют величину Rf — отношение длины пробега анализируемого вещества к длине пробега растворителя (рис. 8). После хроматографирования измеряют расстояние от центра пятна до стартовой линии (отрезок АБ) и от линии фронта жидкости до стартовой точки (отрезок АВ). Отношение отрезков АБ к АВ обозначают величиной Rf:
Rf = АБ / АВ
Величина Rf характерна для данного соединения на данном сорбенте в данной системе растворителей. Она зависит от качества и активности сорбента, толщины слоя, природы растворителей и их соотношения, а поэтому не всегда воспроизводима.
Более надежной оценкой хроматографической подвижности вещества является величина R,. Для ее определения находят величину Rf исследуемого вещества и Rf вещества, принятого за стандарт. Их отношение малочувствительно к влиянию случайных отклонений в условиях эксперимента:
Rs = Rf вещества / Rf стандарта
Поэтому Rs — более воспроизводимая и относительно постоянная величина.

Общий ТСХ-скрининг в нормально-фазовом варианте был разработан на кафедре токсикологической химии ММА им. И.М.Сеченова для систематического судебнохимического анализа биологических объектов на лекарственные и наркотические вещества, включенные по Приказу М3 СССР №1021 в обязательный круг исследования эксперта в случае ненаправленного анализа. Этот метод применим для веществ кислотного, нейтрального, слабоосновного и основного характера. ТСХ-скрининг предполагает разделение веществ в общих системах растворителей на хроматографические зоны с последующим исследованием каждой зоны, в которой были обнаружены те или иные соединения с использованием частных систем растворителей.
Как мы уже отмечали ранее, лекарственные и наркотические вещества из биологических объектов изолируются путем настаивания с подкисленным спиртом (методы Стаса-Отто, Саломатина), нейтральным ацетоном (метод Карташова), подкисленной водой (методы Васильевой, Степанова-Швайковой, Поповой, Крамаренко), подщелоченной водой (метод Валова). Из полученных извлечений (вытяжек) вещества кислотного, нейтрального и слабоосновного характера экстрагируются диэтиловым эфиром или хлороформом при рН=2-2,5, а вещества основного характера — при рН=8-10. Хлороформные (эфирные) экстракты упаривают до небольшого объема и исследуют.
Применительно к веществам, включенным в учебную программу по токсикологической химии, схему ТСХ-скрининга в общих системах растворителей можно представить следующим образом (по Б.Н.Изотову).
ТСХ-скрининг веществ кислотного, нейтрального и слабоосновного характера
Цель: выявить наличие токсикологически важных лекарственных веществ в извлечениях и определить их принадлежность к определенной группе соединений.
Условия анализа: сорбент — закрепленный слой силикагеля. Система растворителей: хлороформ ацетон (9:1). Время насыщения камеры парами системы — 15-20 мин. Длина пробега системы растворителей по пластинке 10 см. На стартовую линию хроматографической пластинки наносят (см. рис. 9):
— полоса А — растворы стандартов (например, барбамила, антипирина);
— полоса Б — 1/25 часть эфирного экстракта, полученного из водного извлечения при рН=2;
— полоса В — стандарт (раствор циклобарбитала);
— полоса Г — 1/10 часть эфирного экстракта, полученного из водного извлечения при рН=2.

Реагенты-проявители для обнаружения веществ на пластинке:
— слой сорбента полос А, Б и В обрабатывают 5% раствором сульфата ртути (II) и 0,1% раствором дифенилкарбазона в хлороформе. Производные барбитуровой кислоты проявляются в виде фиолетовых пятен. Пластинку нагревают горячим феном (~50°С). Пятна обесцвечиваются.
— затем эти же полосы на пластинке обрабатывают 10% раствором хлорида железа (III). При этом проявляются производные пиразола, салициловой и бензойной кислот;
— при последующей обработке этих полос модифицированным реактивом Драгендорфа и 10% раствором серной кислоты обнаруживаются вещества основного характера, такие как кофеин, производные 1,4-бензодиазепина и др. Обработка пластинки раствором серной кислоты повышает чувствительность реактива Драгендорфа.
Все вещества кислотного, нейтрального и слабоосновного характера делятся на 3 хроматографические зоны в зависимости от значения величины Rf в указанной общей системе растворителей.
Зона 1 включает 2 вещества, имеющие значение Rf 0-0,25. В этой зоне обнаруживаются антипирин (Rf 0,19) и кофеин (Rf 0,25).
Зона 2 включает 7 веществ, имеющих значение Rf 0,31-0,41. В этой зоне обнаруживаются фенобарбитал (Rf 0,31), барбитал (Rf 0,32), нитразепам (Rf 0,35), барбамил (Rf 0,37), этаминал-натрий (Rf 0,37), бутобарбитал (Rf 0,41), циклобарбитал (Rf 0,41).
Зона 3 включает вещества, имеющие значение Rf 0,41-0,64. В этой зоне обнаруживается диазепам (Rf 0,62).
Полоса Г на пластинке используется для соскабливания сорбента (заштрихованная часть), элюирования веществ подходящим растворителем (1 зона — метанолом; 2 и 3 зоны — ацетоном) и последующего хроматографического исследования в частных системах растворителей, в которых все вещества обнаруженной группы соединений хорошо разделяются между собой.
ТСХ-скрининг в анализе веществ основного характера
Цель: выявить наличие в извлечении из объекта веществ, являющихся соединениями основного характера.
Условия анализа: сорбент — закрепленный слой силикагеля. Система растворителей: хлороформ — диоксан — ацетон — 25% раствор аммиака (45:47,5:5:2,5), время насыщения камеры парами системы — 15 мин. Пробег растворителя по пластинке — 10 см.
На стартовую линию хроматографической пластинки наносят (рис. 10):
— полоса А — растворы стандартов (например, атропина и новокаина);
— полоса Б — 1/25 часть хлороформного экстракта, полученного из водного извлечения при рН=8-10;
— полоса В — раствор стандарта этаперазина;
— полоса Г — 1/10 часть хлороформного экстракта, полученного из водного извлечения при рН=8-10.

Реагенты-проявители: слой сорбента полос А, Б, В (при закрытой полосе Г) последовательно обрабатывают:
— 10% раствором хлорида железа (III). Возможно обнаружение производных фенотиазина и пиразола;
— 5% раствором нитрита натрия, содержащим 3% хлорной кислоты. Обнаруживают ярко выраженные окрашенные пятна производных фенотиазина;
— 10% раствором серной кислоты и просматривают в УФ-лучах. Возможно обнаружение хинина по голубой флуоресценции;
— модифицированным реактивом Драгендорфа. Вещества основного характера проявляются в виде оранжевых пятен на бледно-желтом фоне.
Соединения основного характера делятся на пластинке на 4 хроматографические зоны, в зависимости от значения Rf.
Зона 1 включает 8 веществ со значением Rf 0,12-0,36. В этой зоне обнаруживаются пахикарпин (Rf 0,12), морфин (Rf 0,14), атропин (Rf 0,15), эфедрин (Rf 0,18), хинин (Rf 0,25), стрихнин (R 0,25), кодеин (Rf 0,28), этилморфин (Rf 0,31).
Зона 2 включает 3 вещества со значением Rf 0,5-0,58. В этой зоне обнаруживаются этаперазин (Rf 0,51), антипирин (Rf 0,54), кофеин (Rf 0,56).
Зона 3 включает 7 веществ со значением R, 0,63-0,83. В этой зоне обнаруживаются хлордиазепоксид (Rf 0,63), дипразин (Rf 0,66), новокаин (Rf 0,7), тиоридазин (Rf 0,72), аминазин (Rf 0,75), промедол (Rf0,76), нитразепам (R, 0,77).
Зона 4 включает 4 вещества со значением Rf 0,87-0,98. В этой зоне обнаруживаются левомепромазин (Rf 0,87), папаверин (Rf 0,91), диазепам (Rf 0,95), кокаин (Rf 0,98).
Полосу Г (заштрихованная часть) соскабливают, элюируют соответствующим растворителем, для хроматографической зоны 1 применяют метанол — диэтиламин (9:1), для 2, 3,4 хроматографических зон метанол — 25% раствор аммиака (9:1). Затем проводят исследование полученных элюатов в частных системах растворителей, в которых вещества данной зоны хорошо разделяются.
Если ни в одной из зон ни с одним из реактивов пятна на пластинках не обнаружены, делают заключение о ненахождении в объекте исследуемых веществ. При обнаружении пятна в какой-либо из зон проводят основное исследование с использованием подтверждающих реакций и методов.
При экспресс-анализе острых отравлений наркотическими и психотропными веществами ТСХ-скрининг проводят на двух пластинках в двух системах растворителей:
1. толуол — ацетон — этанол — 25% раствор аммиака в соотношении компонентов 45:45:7,5:2,5;
2. диоксан — хлороформ — ацетон — 25% раствор аммиака в соотношении компонентов 47,5:45:5:2,5.
На обе пластинки «Силуфол УФ-254» наносят аликвоты извлечений из объекта и стандартные растворы морфина, кокаина, кодеина, аминазина, димедрола, амитрип- тилина и помещают в указанные системы растворителей. Когда системы растворителей достигают высоты 10 см, пластинки вынимают и, после высушивания, просматривают в УФ-свете. Отмечают пятна стандартных веществ и на том же уровне пятна, полученные из вытяжки (извлечения из объекта). Дополнительно на эти зоны наносят капельно реактивы на предполагаемые вещества.
Например:
— смесь серной кислоты и этанола (1:9) для производных фенотиазина;
— реактив Марки для опиатов;
— концентрированную серную кислоту для димедрола и т.д.
Хроматографирование в двух системах растворителей разной полярности позволяет достаточно надежно провести групповое, а в ряде случаев индивидуальное обнаружение наркотических и психотропных веществ.
ТСХ-скрининг токсических веществ кислотного и основного характера по методу В.А.Карташова
Исследование проводят на специально приготовленных хроматографических пластинках по следующей методике. К 3 г силикагеля марки КСК добавляют 0,2 г гипса и 7,5 мл воды для веществ кислотного характера или 0,025 М раствора гидроксида калия для веществ основного характера. Смесь перемешивают в ступке до однородной массы, наносят на обезжиренную стеклянную пластинку размером 9х 12 см, подсушивают и активируют при 120°С 30 мин.
Вещества кислотного характера
Сухой остаток, полученный при изолировании по методу В.А.Карташова и содержащий вещества кислотного характера, растворяют в небольшом объеме хлороформа и количественно наносят на стартовую линию хроматографической пластинки (рис. 11). По краям стартовой линии наносят хлороформные растворы «стандартов» (дифенин и тиопентал).
Условия анализа. Система растворителей: ацетон — н-гексан — диэтиламин (10:10:1). После высушивания пластинку обрабатывают раствором сульфата ртути. Вещества кислотного характера проявляются в виде полосы, а «стандарты» — в виде пятен белого цвета.
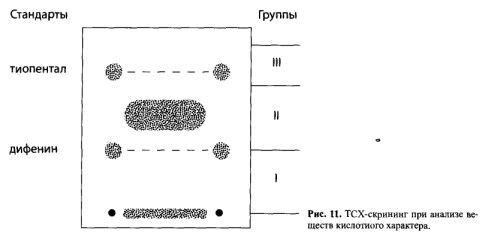
Таблица 8. Хроматографические группы веществ кислотного характера
|
1 группа |
2 группа |
3 группа |
|
Ацетилсалициловая кислота |
Барбамил |
Ноксирон |
|
Салициловая кислота |
Барбитал |
Тиопентал |
|
Бензойная кислота |
Этаминал-нагрий |
Мепробамат |
|
Фенобарбитал |
Дифенин |
Таблица 9. Распределение веществ по хроматографическим группам
|
1 группа 22 вещества |
2 группа 16 веществ |
3 группа 7 веществ |
4 группа 18 веществ |
5 группа 12 веществ |
6 группа 5 веществ |
|
Анабазин |
Аминазин |
Дикаин Скополамин и др. |
Феназон |
Папаверин |
Эфедрин Седуксен Резерпин и др. |
По полученным данным вещество относят к одной из трех хроматографических групп (табл. 8).
Зону силикагеля, содержащую исследуемое вещество, соскабливают, элюируют хлороформом, который далее исследуют с помощью частных реакций и физико-химических методов.
Вещества основного характера
К экстракту из щелочного раствора, полученного при изолировании по методу В.А.Карташова, добавляют 2 капли 10% спиртового раствора хлороводородной кислоты и выпаривают при 40°С досуха. Остаток растворяют в небольшом объеме хлороформа, насыщенного аммиаком, и наносят в виде полосы на стартовую линию хроматографической пластинки (рис. 12). Справа и слева в две точки на линию старта наносят смесь из 5 «стандартов»: кодеина, дикаина, новокаина, амидопирина и седуксена.
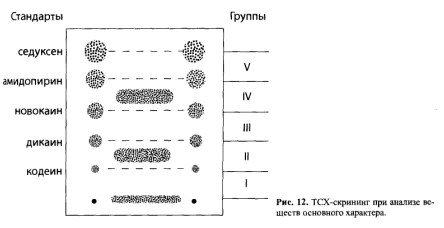
Условия анализа. Подвижная фаза — ацетон. После высушивания пластинку рассматривают в УФ-лучах (светофильтры 254 и 360 нм), отмечают флуоресцирующие зоны, затем обрабатывают реактивом Драгендорфа. «Стандарты» образуют на пластинке пятна оранжевого цвета, и пластинка делится на 6 зон. Исследуемое вещество, проявляющееся; на пластинке в виде окрашенной полосы, попадает в одну из шести хроматографических групп (см. табл. 9).
Если ни в одной из зон не обнаруживаются полосы оранжевого цвета, делают заключение, что в исследуемом объекте вещества основного характера отсутствуют. Если обнаружено пятно, окрашенную зону снимают с пластинки, добавляют гидроксид натрия и экстрагируют диэтиловым эфиром. Эфир испаряют и проводят основное исследование на вещества, относящиеся к определенной группе, используя частные химические реакции и различные физико-химические методы.
7.1.3. ТСХ-скрининг в варианте «Toxi-Lab АВ»
В этом варианте модифицирован классический метод тонкослойной хроматографии. Это специально разработанная аналитическая система, которая предназначена для исследования одной пробы объекта (мочи) на наличие наркотических средств, психотропных и сильнодействующих веществ. Эта система обеспечивает стадию пробоподготовки, экстракцию, концентрирование, разделение веществ и их обнаружение.
Эта система представлена тремя наборами экстракционных пробирок с органическим растворителем определенного состава, набором ТСХ-пластин, концентрационных дисков и сосудами для обнаружения веществ:
— «Toxi-Lab А» используется для обнаружения веществ основного, нейтрального характера (опиаты, метадон, амфетамины и др.);
— «Toxi-Lab В» используется для обнаружения веществ кислотного и нейтрального характера (барбитураты, органические кислоты и др.);
— «Toxi-Lab Cannabinoid» используется для обнаружения тетрагидроканнабинола и его производных.
Методика использования системы включает несколько стадий:
— экстрагирование из мочи в специальных экстракционных пробирках экстрагентом определенного состава при рН=4,5 веществ кислотного и нейтрального характера (барбитураты и др.), при рН=9 веществ основного и нейтрального характера (опиаты, амфетамины, метадон и др.);
— концентрирование полученных экстрактов на сорбирующем материале, представленном в системе в виде микродисков (диаметром 2,5 мм) под струей теплого воздуха.
— хроматографирование с использованием специальных пластин, имеющих углубления на линии старта, в которые вставляются диски с сорбированными веществами (рис. 13). Пластины изготовлены из микроволоконной стеклобумаги, пропитанной кремниевой кислотой и раствором соли ванадия («Toxi-Lab А») и без раствора соли ванадия. («Toxi-Lab В»). Системы растворителей для «Toxi-Lab А»: этилаце- тат — метанол — вода очищенная (29:10:5), для «Toxi-Lab В»: этилацетат — дихлорметан (40:60). К обеим системам добавляют 25% раствор аммиака в отношении 1:4 или 1:5 (зависит от условий лаборатории).
— обнаружение веществ на пластинах проводится путем последовательного погружения их в сосуды, заполненные специальными хромогеннымисосгавами (реактив Марки-Манделина, модифицированный реактив Драгендорфа и др.); также облучение пластинок УФ-светом при 366 или 254 нм.
На рисунке 13 представлены пластины из каталога системы «Toxi-Lab А» для веществ основного и нейтрального характера. Пластины I, II и III детектированы с помощью реактивов Марки-Манделина. Сначала пластинки выдерживали в парах 37% формальдегида и затем погружали в концентрированную серную кислоту, содержащую 0,1% ванадата ммония. Пластина III промыта погружением в воду и освещена УФ-лучами при 254 нм, IV пластина обработана модифицированным реактивом Драгендорфа. На пластины в места 1, 2, 3, 4 фирмой-производителем нанесены растворы стандартных соединений лекарственных и наркотических веществ.
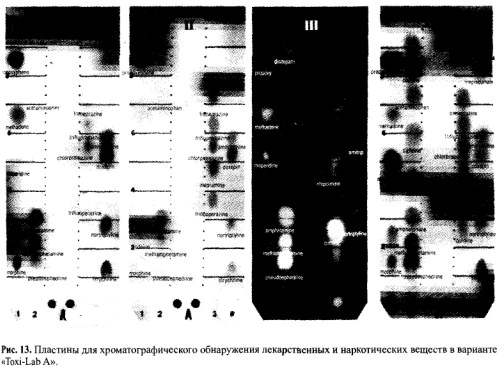
Идентификация веществ на пластинах проводится по окрашенным в определенные цвета или флуоресцирующим пятнам путем сравнения с характеристиками стандартных соединений, нанесенных на пластины производителем.
Метод ТСХ в варианте «Toxi-Lab» позволяет не использовать один из факторов плохой воспроизводимости результатов ТСХ-анализа — нанесение пробы на пластинку в ее классическом варианте. Многостадийное детектирование исключает возможность неправильной интерпретации получаемых результатов. Предел обнаружения веществ составляет 1 мкг/мл (морфин). В наборе реактивов и оборудования в данной системе предусмотрено наличие каталога с образцами результатов анализа более чем для 100 различных токсических веществ и их метаболитов.
7.1.4. ТСХ-скрининг в обращенно-фазовом варианте (ОФТСХ)
Метод разработан сотрудниками кафедры токсикологической химии Пермской государственной фармацевтической академии. Сорбентом является тонкий слой силикагеля «Сорбтон-2» с привитой алкильной фазой С2 и «Плазмохром» с привитой фазой С.. Привитые фазы придают сорбенту гидрофобные свойства. В качестве связующего компонента использована соль кремниевой кислоты, за счет которой слой оказался достаточно прочным и позволил применять для детектирования последовательно несколько реагентов с целью обнаружения различных групп химических соединений. Для веществ кислотного, нейтрального и слабоосновного характера авторами предложена система растворителей этанол — вода (3:7), для веществ основного характера — этанол — вода — 25% раствор аммиака (6:5,5:0,5). Для обнаружения токсических веществ на хроматографических пластинках в качестве реактивов рекомендуются:
— для веществ кислотного характера — растворы хлорида железа (III), растворы сульфата ртути и дифенилкарбазона (в хлороформе);
— для веществ нейтрального характера — фурфурол, подкисленный йодплатинат;
— для веществ основного характера — раствор нингидрина, раствор серной кислота в этаноле, реактивы Драгендорфа, Марки, Манделина и др.
Предел обнаружения веществ на пластинках при обращенно-фазовом варианте в 2-3 раза меньше по сравнению с нормально-фазовым вариантом ТСХ. Идентифицируют вещества на пластинках по величине Rf или по величине R, при хроматографировании в присутствии вещества-«стандарта».
7.1.5. Внутригрупповой ТСХ-скрининг в частных системах растворителей
Любой вариант ТСХ может быть использован для определения индивидуальных веществ в извлечениях из биологического материала. Для этого необходимо использовать условия, разработанные для узкого круга ядовитых веществ. В качестве «стандартов» берут вещества из этой же группы. Как пример приводим определение индивидуального барбитурата, если на предварительном этапе выявлено наличие барбитуратов в извлечении (методика описана Б.Н.Изотовым).
На пластинку со слоем силикагеля, как показано на рисунке 14, наносят растворы стандартов: барбитала, фенобарбитала, барбамила, этаминала, бутобарбитала (по 2 капли 1 % растворов), а рядом — извлечение из объекта или часть элюата, полученного с пластинки (полоса Г на рис. 10).
Для обнаружения места расположения пятен используют специальные реактивы, которыми опрыскивают с помощью пульверизатора пластинку после ее проявления в системе растворителей и высушивания.
Система растворителей (частная для барбитуратов): хлороформ — н-бутанол — 25% раствор аммиака (70:40:5). Сорбент — силикагель КСК, забуференный 0,1 М раствором борной кислоты.
Для производных барбитуровой кислоты пластинку обрабатывают в начале раствором сульфата ртути (II), а затем раствором дифенилкарбазона в хлороформе.
Барбитураты проявляются в виде сине-фиолетовых или красно-фиолетовых пятен и имеют следующие значения Rf: барбитал — 0,55; фенобарбитал — 0,40; барбамил — 0,90; этаминал — 0,95; бутобарбитал — 0,65.

Пятно стандарта и пятно анализируемого вещества должны иметь одинаковую окраску и одинаковое значение Rf.
Можно привести такие же примеры по определению индивидуальных веществ из других трупп токсикологически важных веществ (морфинана, хинолина, фенилалки- ламина и др.).
7.1.6. Газожидкостная хроматография
Газожидкостная хроматография является одним из эффективных методов при проведении скрининга. Основным условием использования этого метода является летучесть соединений при температуре испарителя. Во многих методиках рекомендуется переводить исследуемые вещества в легколетучие соединения путем их дериватизации. В таких случаях метод ГЖХ является универсальным способом для проведения скрининга.
Газожидкостная хроматография — это один из видов распределительной хроматографии. Разделение веществ происходит в специальных колонках, заполненных твердым носителем, представляющим собой пористый материал природного или синтетического происхождения (пемза, кизельгур, полисорб, целит и др.). Зернистый носитель набивается в колонку — трубку малого диаметра длиной от нескольких сантиметров до нескольких метров. Колонки изготовляют из нержавеющей стали, меди, алюминия, стекла и других материалов. Твердый носитель имеет тонкий слой неподвижной фазы.
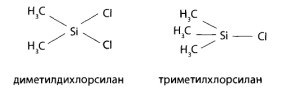
Неподвижная жидкая фаза может быть неполярной — апиезоны (L, М, N, SE-30, OV-1, OV-101). Это углеводороды и полимеры, в основном, диметил- и триметилсилана:
Неподвижная фаза может быть полярной и чаще всего представлена полиэтиленгли- колем с массой 20 ООО — карбовакс-20М, фенилметилсиланом OV-17, цианоэтилсиланом ХЕ-60 и др. Подвижной фазой является инертный газ, чаще всего азот или гелий. Принцип устройства газожидкостного хроматографа приведен на рисунке 15. Исследуемый объект вносят в дозатор (3) в количестве нескольких микролитров с помощью микрошприца. Вещества переносятся газом-носителем (7) в колонку (5). На колонке происходит разделение компонентов смеси. Разделение смеси зависит от величины коэффициентов распределения веществ между подвижной и неподвижной фазами и от эффективности колонки. Эффективность колонки выражается числом теоретических тарелок. Под «теоретическими тарелками» понимают количество теоретических ступеней, на которых устанавливается равновесие между подвижной и неподвижной фазами. Чем больше таких ступеней на единицу длины колонки, тем лучше происходит разделение веществ.
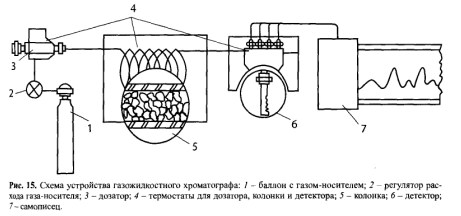
Из колонки разделенные вещества поступают в детектор (6). Детектор — часть прибора, которая регистрирует интенсивность определенных свойств бинарных смесей (компонент + газ-носитель). Изменение интенсивности сигнала детектора свидетельствует об изменении состава газовой смеси. Детекторы могут быть настроены на измерение интенсивности различных физико-химических свойств системы (плотности, теплопроводности, теплоты сгорания, ионизации и т.д.), которые преобразуются в электрический сигнал детектора, регистрируемый самописцем (7).
В практике химико-токсикологического анализа используют катарометр — детектор теплопроводности. Катарометром измеряют разность теплопроводностей чистого газа- носителя и смеси газа-носителя с анализируемым веществом. Чувствительность определения токсических веществ с помощью катарометра находится в пределах 10-3— 10-5 г.
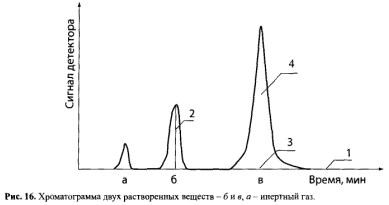
Пламенно-ионизационный детектор (ПИД). Принцип действия основан на измерении тока, возникающего при ионизации молекул органических веществ в пламени водорода. При горении чистого водорода ионы почти не образуются, поэтому электропроводность водородного пламени очень низка. Органические вещества, сгорающие в пламени водорода, образуют ионы или радикалы. Появление заряженных частиц обусловливает электропроводность пламени. Увеличение электропроводности повышает силу ионного тока, которая отображается на хроматограмме в виде пика. Чувствительность ПИД — 10-9 — 10-12 г. Хроматограмму двух растворенных веществ можно представить следующим образом (рис. 16).
На кривой разделения каждому пику свойственны следующие параметры. Высота пика (2) — это расстояние от вершины пика до его основания (3). Площадь пит (4) — площадь, заключенная между контуром пика и его основанием. Основание пика (3) — отрезок нулевой линии (1) между крайними точками пика.
Качественной характеристикой в газожидкостной хроматографии считаются:
— Время удерживания вещества. Это время от момента введения смеси веществ в дозатор до момента вычерчивания максимума пика самописцем. При постоянных условиях анализа характерно для данного вещества.
— Относительное время удерживания. Это отношение времени удерживания исследуемого вещества к времени удерживания стандарта.
— Объем удерживания. Эту величину получают путем умножения времени удерживания на объемную скорость газа-носителя. Это общий объем подвижной фазы, необходимый для удаления данного вещества из колонки.
— Относительный объем удерживания. Его определяют в присутствии стандартного вещества путем деления найденного времени удерживания компонентов на время удерживания стандартного вещества после вычитания времени удерживания инертного газа-носителя из обоих значений времени удерживания.
— Удельный удерживаемый объем. Это объем удерживания на 1 г неподвижной жидкой фазы. Эта величина является константой, подобной точке кипения, и более надежна для идентификации.
— Индекс удерживания (I). Индекс удерживания предложен Ковачем в 1958 г. Он основан на логарифмической шкале, в которой нормальные парафины имеют значения индексов удерживания, в 100 раз превышающие число атомов углерода в их молекуле. Например, 200, 300, 400 для этана, пропана, н-бутана соответственно. Поэтому величины 1/100 рассматривают как число атомов углерода в н-парафине, который определялся бы с интересующим нас веществом. Индекс удерживания — более воспроизводимая характеристика для вещества, чем относительный объем удерживания. Он пропорционален логарифму удельного объема удерживания.
В основе идентификации веществ с помощью ГЖХ — сравнение индекса удерживания неизвестного вещества с индексом удерживания известного соединения.
7.1.6.1. ГЖХ-скрининг в анализе лекарственных и наркотических веществ в извлечениях из мочи
Исследуемые вещества выделяют из мочи путем экстракции или сорбции. Для ГЖХ- скршшнга достаточно использовать часть извлечения из объекта, которая соответствует по объему 10 мл мочи (представлено по методикам, описанным С.К.Ереминым).
Условия хроматографического анализа: прибор ЛMХ-80, детектор термоаэрозольный (ТАД), термоионный или беспламенный азотно-фосфорный (NPD), колонка стеклянная, силанизированная, длиной 1 м, внутренний диаметр — 2-3 мм, сорбент — 3% SE-30 на хромосорбе W (HP) — 80-100 меш., скорость газа-носителя (азота) для ТАД 45 мл/мин и для NPD — 40 мл/мин (гелия), эффективность колонки 1200 т.т. (ТАД) и 1300 (NPD), температура детектора — 300°С, температура испарителя 250°С, температура испарителя по линейной программе от 130 до 290°С, объем пробы — 2,5 мкл.
Для идентификации хроматографических пиков определяют индекс удерживания (Ix). С этой целью используют в качестве стандартов смесь н-алканов СnН2n+2 с числом атомов углерода от 10 до 32, которым присвоены значения индексов, равные 100n (n — число атомов углерода в алкане). Расчет проводят по формуле:
Ix = 100n + (In+1 – In) · [(lg tx – lg tn) / (lg tn+3 – lg tn)]
где Ix — индекс удерживания анализируемого вещества, tx, to, tn+1— исправленное время удерживания неизвестного вещества (tx) и ближайших к нему реперных н-алканов с числом атомов углерода n (tn) и n+1(tn+1), In+1 и In — индексы удерживания н-алканов.
Вместо исправленного времени удерживания иногда используют в расчетах истинные удерживаемые объемы.
При работе с другими детекторами вместо н-алканов используют в качестве стандартов смесь азотсодержащих веществ, индексы удерживания которых также предварительно определены по н-алканам при тех же условиях (см. табл. 10). Стандартное отклонение измеренных индексов удерживания находится в пределах 15-20 единиц. При скрининге «поисковое окно» должно находиться в пределах ±50-60 единиц I, если сравнивается I неизвестного соединения с I известных веществ.
Таблица 10. Смесь анализируемых веществ для расчета индексов удерживания
|
Вещество |
Индекс удерживании по н-алканам |
|
Амитриптилин |
2193 |
|
Барбитал |
1490 |
|
Кодеин |
2382 |
|
Метиламфетамин |
1220 |
|
Новокаин |
2022 |
|
Папаверин |
2848 |
|
Промедол |
1838 |
|
Стрихнин |
3110 |
Индексы удерживания зависят от концентрации исследуемого соединения в растворе. Многие лекарственные вещества, которые не относятся к списку наркотических и одурманивающих, имеют индексы удерживания, близкие к анализируемым соединениям, и могут влиять на результаты идентификации. Поэтому ГЖХ-скрининг используют для отсева отрицательных проб и предварительного обнаружения веществ различных фармакологических групп. Все пробы, давшие положительный результат, подтверждаются другими надежными методами.
7.1.6.2. ГЖХ-скрининг в анализе «летучих» ядов
Методика газожидкостной хроматографии «летучих» ядов основана на исследовании не самой биологической жидкости (крови, мочи), а газовой фазы, находящейся над ней. Этот способ назван парофазным анализом (ПФА) или анализом равновесного пара. В его основе — один из законов Д.П.Коновалова, выражающий зависимость состава пара от состава раствора: «повышение относительного содержания компонента в жидкой фазе всегда вызывает увеличение относительного содержания его и в парах». ПФА основан на технике и принципе газовой экстракции. Простейший вариант ПФА: в герметично закрывающийся флакон объемом V помещают исследуемый объект объемом V1, содержащий определенный «летучий» яд с концентрацией С1. Объем газовой фазы над объектом Vg равен V-V1. Систему выдерживают при постоянной температуре до установления термодинамического равновесия (равновесного распределения «летучего» яда между жидкостью и газом). Газовую фазу вводят в хроматограф. Измеряют абсолютное значение равновесной концентрации яда Cg. Поскольку в процессе установления фазового равновесия некоторая часть «летучего» яда переходит в газовую фазу, его концентрация в конденсируемой фазе будет меньше исходной С1. Количество вещества, перешедшего в газовую фазу из раствора, зависит от соотношения объемов фаз R=Vg/V1 и коэффициента распределения K=C1/Cg. Если пренебречь изменением объема жидкости за счет испарения раствора в процессе установления равновесия, концентрацию вещества в исходном растворе можно вычислить по его содержанию в равновесной газовой фазе по уравнению C1=Cg (K+R). Эта формула лежит в основе парофазного анализа.Метод применяют для определения летучих органических веществ в питьевой воде, природных и сточных водах, пищевых продуктах, крови, моче, фармацевтических и косметических препаратах, а также в криминалистике, микробиологии, медицинской диагностике и др. В настоящее время имеются специальные автоматические анализаторы и приставки к газовым хроматографам, позволяющие проводить парофазный анализ.В анализе летучих соединений Международный совет по систематическому химико-токсикологическому анализу рекомендует следующие условия проведения ГЖХ- скрининга.
Материал колонки: хромосорб W, фр. (80-100 меш.); покрытие (неподвижная фаза) — 15% карбовакс 1500, температура: изотермический режим (60-100°С); тестовая смесь (1 г/л): метанол, ацетон, этанол, изопропанол, используемые с целью проверки качества колонки, чувствительности детектора и эффективности разделения; детектор: пламенноионизационный (ПИД).
Методика исследования биологических объектов с помощью парофазного анализа при проведении ненаправленного анализа сводится к следующему.
Внутренние органы и ткани. Навеску измельченного объекта массой 5 г помещают в соответствующую емкость объемом 15 мл, добавляют 0,5 мл 0,5% раствора фосфорновольфрамовой кислоты. Емкость плотно закрывают резиновой пробкой, обкатывают алюминиевым колпачком и нагревают на кипящей водяной бане не менее 15 мин.
Кровь, моча. Исследуемый объект объемом 0,5 мл помещают в емкость объемом 15 мл, закрывают резиновой пробкой, обкатывают алюминиевым колпачком и оставляют при комнатной температуре на 5-10 мин. В системе устанавливается термодинамическое равновесие между жидкой и паровой фазами.
Затем в емкостях прокалывают иглой медицинского шприца резиновую пробку, отбирают 2 мл парогазовой фазы и вводят в испаритель газового хроматографа.
Для обнаружения и определения этанола и других летучих спиртов применяется утвержденный экспрессный этилнитритный метод, который позволяет обнаружить и определить алифатические спирты С,-С5 в виде алкилнитритов (метод описан в разделе 9.1).
Остальные «летучие» яды в связи с плохим разделением определяют, используя две параллельные колонки с селективными неподвижными фазами различной полярности. Селективность метода повышается модификацией твердого носителя слоем металлического серебра.
Условия анализа: прибор ЛXM-80 или «Цвет», детектор ПИД, скорость потока водорода 27-30 мл/мин, воздуха — 200-360 мл/мин, газ-носитель — гелий (24 мл/мин) или азот (30 мл/мин), колонки металлические (1.-1 -2 м, с1м=0,3 см), температура колонки — 80- 85°С, испарителя — 100-110°С, твердый носитель — целит С-22 (фракция 60-80 меш.), модифицированный слоем металлического серебра, или хроматон AW DMCS (0,20-0,25 мм), неподвижные жидкие фазы: 1,2,3-трис (2-цианоэтокси)пропан или карбовакс 15% 20 М (1-я колонка) и тритон Х-100 или 10% дионилфталат (2-я колонка).
Методика. В два пенициллиновых флакона емкостью 10 мл вносят по 1 мл 10% раствора фосфорно-вольфрамовой кислоты и по 1 мл исследуемой пробы. Содержимое флаконов перемешивают и вносят в каждый флакон по 2,5 г безводного сульфата натрия. Флаконы закрывают резиновыми пробками и обкатывают алюминиевыми колпачками. Один флакон помещают на 5 мин в кипящую водяную баню, взбалтывая сразу и затем через 3 минуты. Из этого флакона медицинским шприцем отбирают 0,5 мл парогазовой фазы и вводят ее в 1-ю колонку. Через 5 мин второй флакон помещают в водяную баню и 0,5 мл парогазовой фазы вводят во 2-ю колонку.
По полученным пикам на первой колонке рассчитывают индексы удерживания или tотн и сравнивают с табличными данными. Выбирают вещества, у которых индексы удерживания (tотн) совпадают или отличаются в пределах «поискового окна». Отобранные вещества включают в круг предполагаемых соединений. Остальные вещества исключаются из дальнейших исследований, что сужает круг поиска.
На второй колонке происходит разделение отобранных веществ. Порядок их выхода из колонки также изменяется. Далее рассчитывают индексы удерживания (tотн) и сравнивают их с индексами удерживания (tотн) стандартных веществ из числа предполагаемых ядовитых соединений. Анализируя данные, полученные на 2 колонках с неподвижными фазами разной полярности, делают вывод о присутствии тех или иных «летучих» ядм в исследуемом объекте.
Результаты подтверждаются химическим методом. Если при проведении газохрс- матографического скрининга получен отрицательный результат, т.е. на хроматограмма не обнаружено пиков, характеризующих присутствие какого-либо ядовитого вещества делают вывод о ненахождении группы «летучих» ядов в объекте.
7.1.7. Иммунохимические методы скрининга лекарственных и наркотических веществ
Этот раздел представлен с использованием разработок С. К. Еремина.
Иммунохимические методы (скрининг-тесты), используемые для анализа наркотических, психотропных и одурманивающих веществ в биологических жидкостях (крови, моче, слюне) являются высокочувствительными. Они позволяют за минимальное время из достаточно большого круга исследуемых соединений выявить одно или несколько веществ. Скрининг-тесты не отличаются высокой специфичностью и часто вообще неспецифичны. Особенность этих методов в том, что при их использовании нет необходимости проводить изолирование веществ из объекта и применять специальные приемы по их очистке. В основе иммунохимических методов — взаимодействие специфических белковых антител (антисывороток) с анализируемым веществом, выступающим в роли антигена (гаптена). Чем больше концентрация в объекте вещества-антигена, тем больше образуется комплекса антиген-антитело. Чтобы детектировать получаемый результат, один из компонентов реакции — гаптен или антитело — метят специальной меткой.
В основу классификации иммунохимических методов положены тип применяемой метки и способ ее детектирования (табл. 11).
Все методы иммунохимического исследования включают 4 этапа.
1 этап. Нанесение метки на препарат или его аналог.
2 этап. Добавление антисыворотки с антителами и образование комплекса антителомеченый препарат.
3 этап. Добавление исследуемой крови (сыворотки) или мочи. Происходит вытеснение анализируемым веществом меченого препарата из комплекса и образование нового комплекса: анализируемое вещество-антитело.
4 этап. Измерение радиоактивности, интенсивности флуоресценции, активности фермента и т.д.
В токсикологической химии для проведения скрининга применяются иммунофер- ментный анализ, радиоиммунный и поляризационный флуороиммуноанализ.
Используют два вида иммунохимических методов анализа:
— анализ гомогенный, в котором все компоненты находятся в растворе;
— анализ гетерогенный, в котором реагирующие компоненты разделяют путем включения стадии «отмывки» или центрифугирования.
Гомогенный иммуноферментный анализ (ИФА)
В гомогенном ИФА в качестве метки используют оксидазы. Среди них чаще всего применяют лизоцим, глюкозо-6-фосфатдегидрогеназу, малатдегидрогеназу. Эти ферменты способны окислять хромогенный субстрат (специальное химическое вещество, например хлорнафтол) с образованием окрашенного соединения.
Таблица 11. Классификация иммунохимических методов анализа
|
Название метода |
Способ детектирования |
|
Радиоиммунный анализ (РИА) |
Радиоактивность |
|
Иммуноферментный анализ (ИФА) |
Ферментная активность |
|
Поляризационный флуороиммуноанализ (ПФИА) |
Интенсивность флуоресцентной поляризации |
|
Люминесцентный иммуноанализ (ЛИА) |
Интенсивность люминесценции |
Если в исследуемой биологической жидкости присутствует наркотическое, психотропное или одурманивающее вещество, анализ пройдет по нижеприведенной схеме.
При отсутствии в биологической жидкости подозреваемого вещества окраски раствора наблюдаться не будет, так как комплекс фермент-препарат останется связанным с антителом и не проявит окислительных свойств. Гомогенный анализ экспрессный, время его проведения I 30 мин (с учетом времени на обработку результатов). Предел обнаружения исследуемых веществ 10 4-10 6 г/мл.
ИФА используется для предварительного установления факта употребления одурманивающих средств. Используемые в анализе антитела — это иммуноглобулины, продуцируемые лимфоцитами животных (мелкого и крупного рогатого скота) при введении им инородных веществ.
Иммуноферментный метод анализа (ИФА) лекарственных и наркотических веществ можно представить следующей схемой:

Полученные антитела будут игнорировать многие соединения, которые не похожи на антигены (гаптены). Однако если в исследуемой пробе будут содержаться структурнородственные соединения, антитела их также будут связывать. Связывание структурнородственных соединений называется перекрестной реакцией или перекрестной "реактивностью. Среди них некоторые продукты распада белковых молекул (тирании, фенил- пропаноламин), метаболиты ядовитых соединений, лекарственные вещества. Наличие перекрестных реакций с другими лекарственными соединениями может стать причиной ложноположительных реакций.
ИФА всегда используется в комбинации с другими подтверждающими способами и реакциями. При отрицательном результате ИФА не требуется проведения до- полнительных исследований и дается заключение о необнаружении предполагаема веществ.
ИФА в экспресс-диагностике объектов на наркотические и одурманивающие вещества
Этот вариант анализа может проводиться в нелабораторных («полевых») условиях. Используются индикаторные полоски из целлюлозной хроматографической бумаги. Антитела и фермент глюкозооксидазу ковалентно связывают с поверхностью индикаторной полоски. При погружении индикаторной полоски в исследуемый образец содержащееся в нем наркотическое вещество связывает определенную часть активных центров антител, находящихся на индикаторной полоске. Затем эту полоску переносят в раствор смеси, состоящий из определяемого наркотика, пероксидазы, глюкозы и хромогенного субстрата. Определяемое наркотическое вещество на индикаторной полоске связывает активные вакантные центры антител. Глюкоза образует пероксид водорода при взаимодействии с иммобилизированной на индикаторной полоске глюкозоокси- дазой. Пероксид водорода окисляет хромогенный субстрат с образованием окрашенного и нерастворимого продукта. Концентрация определяемого вещества (антигена] обратно пропорциональна интенсивности образующейся окраски. Чем ярче окраска, тем меньше количество вещества в анализируемом растворе. Этот вариант ИФА прост в выполнении и используется в отделениях милиции, спецприемниках, наркологических диспансерах и т. д.
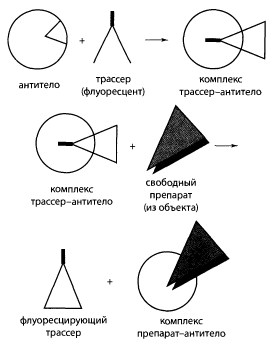
Поляризационный флуороиммуноанализ (ПФИА)
Этот метод используется при анализе уровня лекарственных и наркотических веществ в биологических жидкостях. В качестве меченого препарата применяют молекулу подозреваемого вещества с присоединенной к ней группой, придающей меченой молекуле способность флуоресцировать. В качестве метки используют флуоресцеин. Метод основан на сдвиге угла испускаемого луча света флуоресцирующим образцом при облучении его монохроматическим поляризованным светом. Сдвиг угла испускаемого света при этом сильно зависит от структуры лекарственного или наркотического вещества. Это позволяет определять долю меченых молекул, вытесненных немеченым препаратом, содержащимся в анализируемой пробе (крови, моче). Проведя калибровку системы, можно найти концентрацию препарата в исследуемой пробе.
Этот метод предложен для работы фирмой Эббот (торговая марка ТДХ-анализаторов). Эта фирма выпускает наборы необходимых реагентов для анализа основных противосудорожных, антиаритмических, наркотических и других препаратов из групп опиатов, барбитуратов, производных 1,4-бензодиазепина, каннабиноидов, амфетаминов, кокаина, эфедрина и др. Используемая методика выполняется в автоматическом режиме, отличается высокой чувствительностью и относительной специфичностью.
Поляризационно-флуоресцентный иммуноанализ можно представить следующей схемой:
Гетерогенный иммуноанализ
В качестве метки в этом варианте иммунохимического метода используют ферменты пероксидазу, р-галактозидазу, фосфатазу и др. Метод отличается высокой чувствительностью и позволяет определять вещества в концентрации 10-6 — 10-8 г/л. Время анализа составляет 2-4 ч. Выпускаются диагностикумы для обнаружения алкалоидов группы опия, производных барбитуровой кислоты, эфедрона, каннабиноидов и др.
Метод заключается в том, что препарат (антиген) реагирует с избытком фермент — меченых антител. После образования комплекса антиген-антитело избыток антител связывают добавлением специфического твердофазного иммуносорбента и удаляют отмыванием или центрифугированием. Затем добавляют хромогенный субстрат. Он взаимодействует с фермент-меткой, связанной с антителом, закрепленным на твердом носителе, и образует соответствующую окраску. Если концентрация определяемого вещества в пробе будет значительно превышать концентрацию гаптена, меченого ферментом, то после удаления последнего из реакционной смеси (при отмывании или центрифугировании) хромогенный субстрат не образует окраски (положительный результат анализа). При возникновении окраски результат анализа отрицательный.
Радиоиммунный метод
Этот метод относится к числу гетерогенных методов, так как для анализа степени вытеснения меченого антигена из комплекса антиген-антитело необходимо отделить этот комплекс от раствора. В данном методе используют препарат (или его близкую модификацию), меченый радиоактивным изотопом. С этой целью применяют йод-125 (125I) и радиоактивный изотоп водорода — тритий (3Н), иногда селен. К специфическому антителу (А) добавляют препарат, меченый радиоактивной меткой (R), а затем исследуемый раствор (мочу, плазму крови), содержащий исследуемое вещество (II), которое вытесняет меченый препарат из комплекса.
Определение веществ идет по схеме:
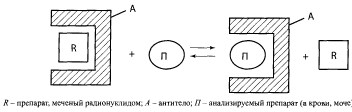
Освободившийся меченый препарат отделяют от раствора и с помощью специальных приборов определяют степень радиоактивности. После определения радиоактивности можно рассчитать концентрацию препарата в растворе. Метод рекомендуется для обнаружения и определения многих лекарственных и ядовитых веществ в крови и моче (психотропных, гормональных, сердечно-сосудистых, производных барбитуровой кислоты, производных 1,4-бензодиазепина, антидепрессантов, наркотических анальгетиков мор- финанового ряда, многих наркотических средств и др.). Этим методом возможно определение веществ в концентрации в 5-15 раз ниже терапевтического уровня. Метод обладаег достаточной специфичностью. Однако она не полная, так как присутствие в биологической жидкости метаболитов препарата или других лекарств, близких по структуре к анализируемому веществу, может привести к ложноположительным результатам и завысить получаемые данные. Методу придается значение при отрицательном результате. Если анализ дает положительный результат, данные подтверждаются другими методами и реакциями.
7.1.8. Аналитический скрининг с помощью химических реакций
На первой стадии анализа необходимо выбрать реакции, позволяющие установить или исключить наличие отдельных групп химических соединений или индивидуальных веществ. Поэтому этот анализ носит также характер скрининга. На этой стадии химикотоксикологического анализа используют наиболее чувствительные реакции. Как правило, эти реакции неспецифичны для отдельных веществ. При отсутствии аналитического эффекта вся группа или конкретное вещество исключается из анализа.
Примеры:
При исследовании извлечений из объекта на вещества основного характера рекомендуется использовать осадительные (общеалкалоидные) реактивы. В анализе применяют не менее 4—5 реактивов, отличающихся наибольшей чувствительностью по отношению к большинству алкалоидов и органических оснований:
— фосфорно-вольфрамовая кислота — H3PO4 · 12WO3 · 2H2O;
— фосфорно-молибденовая кислота — Н3РO4 · 12МоO3 · 2Н2O;
— реактив Драгендорфа — раствор йодида висмута в растворе йодида калия;
— реактив Бушарда — раствор йода в растворе йодида калия;
— реактив Майера — раствор йодида ртути в растворе йодида калия.
Для проведения реакций определенную часть хлороформного экстракта испаряют, добавляют хлороводородную кислоту для переведения токсических веществ в соли и прибавляют 1-2 капли соответствующего реактива.
Если ни с одним из реактивов ни мути, ни осадка не образуется, делают заключение о необнаружении алкалоидов и веществ основного характера. Если хотя бы с одним из реактивов образуется муть или осадок — проводят дальнейшее исследование с использованием подтверждающих методов и реакций.
При анализе извлечений из биологических объектов на группу опийных алкалоидов рекомендованы реакции со специальными реактивами. Для предварительной идентификации опийных алкалоидов и промедола чаще всего используют:
— реактив Марки — раствор формальдегида в концентрированной серной кислоте;
— реактив Фреде — раствор молибдата аммония в концентрированной серной кислоте;
— реактив Эрдмана — смесь концентрированных серной и азотной кислот;
— реактив Манделина — раствор ванадата аммония в концентрированной серной кислоте.
Для проведения реакций на фарфоровых чашках испаряют извлечение из объекта (хлороформный экстракт, полученный из раствора с рН=8-10) и на сухие остатки наносят соответствующие реактивы. При отсутствии окрашивания делают заключение о необнаружении алкалоидов опия и промедола.
Исследование эфирных экстрактов из объектов на производные барбитуровой кислоты проводят, используя реакцию с аммиачным раствором ацетата кобальта. При отсутствии окрашивания дают заключение о необнаружении в исследуемом объекте барбитуратов.
При анализе дистиллята на ядовитые алкилгалогениды используют предварительные реакции отщепления органически связанного хлора и образования изонитрила. При получении отрицательного результата обеих реакций дают заключение о необнаружении в исследуемом объекте хлороформа, хлоралгидрата и четыреххлористого углерода.
Анализ дистиллята на присутствие фенола проводят по реакции с бромной водой, и при отсутствии осадка дается заключение о необнаружении в объекте фенола и кре- золов.
Определение этилового спирта проводят по реакции образования йодоформа. При отсутствии запаха йодоформа и осадка с характерной формой кристаллов дают заключение о необнаружении в объекте этилового спирта
При проведении холинэстеразной пробы в извлечении из объекта на фосфорсодержащие ядохимикаты и получении отрицательного результата дают заключение о необнаружении всей группы фосфорорганических соединений в объекте исследования и т.д.
Скрининговые методы и аналитические реакции предварительного анализа позволяют значительно сократить время, затрачиваемое на исследование биологического объекта на различные группы ядовитых веществ.
7.2. Методы подтверждающего анализа
7.2.1. Метод высокоэффективной жидкостной хроматографии (ВЭЖХ)
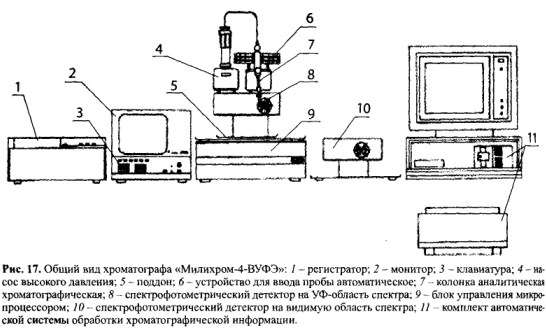
Высокоэффективная жидкостная хроматография является вариантом колоночной хроматографии. Ее отличие от классической колоночной хроматографии заключается в использовании сорбентов с размером зерен 10-30 мкм, поверхностно- и объемно-пористых сорбентов с размером зерен 5-10 мкм, нагнетательных насосов и высокочувствительных детекторов. Быстрый массоперенос при высокой эффективности разделения позволяет использовать ВЭЖХ для разделения и определения веществ в молекулярном или ионном виде. В ВЭЖХ сорбент в колонке пропитан неподвижной фазой (НФ). Подвижной фазой (ПФ) является элюент, который подается в колонку насосом. Элюент движется под давлением 200 и более атмосфер. На рисунке 17 представлен общий вид хроматографа «Милихром-4».
Разделение основано на различной скорости распределения веществ между неподвижной и подвижной фазами. Подвижная фаза подбирается так, чтобы обеспечить хорошее разделение веществ при сравнительно небольшом времени удерживания.
Если состав подвижной фазы в процессе анализа не меняется, этот режим называют изократическим, если соотношение компонентов меняется — градиентным.
В химико-токсикологическом анализе чаще всего используют вариант обращенно- фазовой хроматографии, когда подвижная фаза более полярна, чем неподвижная фаза. Применяются колонки с привитыми фазами. В качестве подвижных фаз рекомендуются смеси вода — метанол, вода — ацетонитрил или буферные растворы, кислоты, основания, В качестве неподвижной фазы чаще всего применяют гидрофобные вещества, привитые на силикагель (например, «Сепарон-18»).
Колонку в приборе ВЭЖХ изготовляют из стали или стекла диаметром 0,3-0,8 см, длиной 6-25 см. Она помещается в термостат, в котором поддерживается определенная температура.
Детектор в ВЭЖХ обычно спектрофотометрический. Он регистрирует оптическую плотность раствора исследуемого вещества при заданной длине волны. Возможно детектирование при нескольких длинах волн.
Идентификация веществ в извлечениях из биологических проб проводится следующим образом:
— сопоставляют время (объем) удерживания определяемого вещества и образца сравнения;
— сравнивают и сопоставляют светопоглощение предполагаемого компонента и образца сравнения при двух или нескольких длинах волн и оценивают их спектральные отношения;
— оценивают совпадение значений времени удерживания определяемого компонента и образца сравнения при добавлении его к экстракту из объекта.
Метод ВЭЖХ нашел широкое применение в настоящее время в практике судебнохимического, клинического анализа при определении и обнаружении в биологических объектах токсических веществ кислотного и основного характера.
Метод ВЭЖХ для общего скрининга не применяется. Это связано с тем, что для каждой группы веществ необходимы специфические условия разделения. Этот метод является оптимальным при частном скрининге, когда круг предполагаемых веществ ограничен (например, при экспертизе наркотического, токсикоманического опьянения).
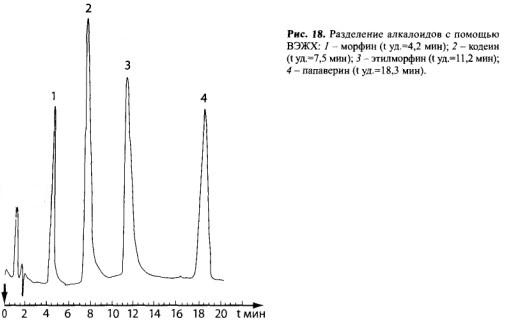
Исследуемые вещества (барбитураты, алкалоиды опия и др.) экстрагируют из подготовленного объекта хлороформом, диэтиловым эфиром или смесью хлороформа и н-бутанола. Органический растворитель испаряют в токе теплого воздуха и растворяют в 100 мкл подвижной фазы, а затем анализируют в приборе, используя условия, рекомендуемые для данной группы соединений.
Для разделения опийных алкалоидов (по методике С. К. Еремина), выделенных из мочи после ее пробоподготовки с помощью кислотного гидролиза, рекомендуются следующие условия:
— жидкостный хроматограф «Милихром-2, -4 или -5»;
— хроматографическая колонка (64×2 мм), заполненная обращенно-фазовым сорбентом марки Сепарон-С18, 5 мкм;
— подвижная фаза (элюент) — смесь 0,01 М водного раствора ацетата аммония (ЧДА, ГОСТ 3117-78) и ацетонитрила (для жидкостной хроматографии) 65:35.
— скорость потока элюента — 100 мкл/мин;
— детектирование проводится при длине волны 230 нм (рис. 18).
7.2.2. Методы ИК- и УФ-спектроскопии
Теоретические основы спектрометрии хорошо изложены в курсе аналитической химии. Эти методы основаны на поглощении электромагнитного излучения определяемым веществом. Область спектра характеризуется определенным участком длин волн электромагнитного излучения (см. табл. 12).
Таблица 12. Спектральные области поглощения
|
Область |
Ультрафиолетовая |
Видимая |
Инфракрасная область |
||
|
ближняя |
средняя |
дальняя |
|||
|
Длина волны, нм |
185-380 нм |
380-750 нм |
750 — 2500 нм |
2500 — 25 000 нм |
— |
|
Волновое |
— |
— |
13300 — 4000 |
4000 — 400 |
400-10 |
|
Тип поглощения |
Электронное |
Вращательное, колебательное, деформационное |
Молекулярное вращение |
||
Для обнаружения ядовитых веществ в токсикологической химии используют спектрометрию в ближней ультрафиолетовой области от 200 до 380 нм, в видимой области; и средней инфракрасной области спектра. Ультрафиолетовая область до 200 нм требует специальных (вакуумных) устройств. Аппаратура для дальней инфракрасной области в нашей стране используется в научных целях и в широкой практике не применяется.
Инфракрасная спектроскопия
Спектр поглощения в инфракрасной области представляет собой сложную кривую с большим числом максимумов и минимумов. Спектральные характеристики (положение максимумов полос, их полуширина, интенсивность) индивидуальной молекулы зависят от масс составляющих ее атомов, геометрического строения, особенностей межатомных сил, распределения заряда и др. Поэтому ИК-спектры строго индивидуальны, что особенно ценно для идентификации вещества. В фармацевтическом и химикотоксикологическом анализе используется область электромагнитного спектра, которая охватывает интервал 4000-250 см '. Совокупность всех полос поглощения, образующая ИК-спектр данного соединения, однозначно определяет его индивидуальность, используется для определения подлинности лекарственного (токсического) вещества и подтверждает его нахождение в извлечениях из объектов химико-токсикологического анализа.
При оценке полученных спектров извлечений из объектов важно знать, какие групповые частоты связаны с наличием в молекуле исследуемого соединения определенных функциональных групп. Такие частоты называются характеристическими. Различные молекулы, содержащие одну и ту же атомную группировку, будут давать в ИК-спектре полосы поглощения в области одной и той же характеристической частоты. Это является основой качественного анализа по ИК-спектрам. Например, полосы в области 3000 — 3600 см-1 могут быть отнесены только О-Н- или N-H-связям, и отсутствие полос в этой области спектра однозначно свидетельствует об отсутствии ОН- и NH-групп в анализируемом веществе. Некоторые полосы приведены в таблице 13.
Таблица 13. Полосы поглощения некоторых функциональных групп
|
Функциональная группа |
Частота, см интенсивность |
|
О-Н |
3650-3200 (переменная) |
|
N-H |
3500-2900 (средняя) |
|
C-H |
3300-2700 (сильная) |
|
C-C |
~2500 (слабая) |
|
C=N |
~2200 (средняя) |
|
C=O |
1850-1650 (сильная) |
|
C=C |
~1650 (средняя) |
|
С-O |
1300-1000 (сильная) |
ИК-спектроскопия используется для обнаружения многих органических соединений, имеющих токсикологическое значение. Методика обнаружения веществ кислотного и основного характера сводится к следующему: сухой остаток после испарения органического растворителя (экстракта из биологического объекта) растирают с сухим мелкоизмельченным бромидом калия в соотношении 1:200 или 1:300 (зависит от марки прибора). Часть смеси переносят в специальную матрицу и прессуют. Полученный прозрачный диск помещают в прибор ИК-спектрофотометр и проводят измерения.
Параллельно анализируют стандартный образец. Совпадение полос поглощения в обоих спектрах свидетельствует об идентичности веществ (табл. 14).
Таблица 14. Основные полосы поглощения некоторых токсических веществ
|
Анализируемое вещество |
Основные пики в ИК-области, см 1 |
|
Барбамил |
1716, 1689,1745 |
|
Кофеин |
1658, 1695, 745 |
|
Феназон |
1660, 770, 1486 |
|
Морфин |
805, 1243, 1448, 945, 1086, 833 |
|
Папаверин |
1507, 1068, 1273 |
|
Атропин |
1720, 1035, 1153 |
|
Стрихнин |
1664, 764, 1392, 1480 |
|
Хинин |
1235, 1510, 1030, 1619 |
|
Эфедрин |
699, 754, 760, 994, 1049, 1242, 1400, 1480, 1605 |
|
Аминазин |
1455, 747, 1240, 1402, 1561 |
|
Новокаин |
1274, 1690, 1605 |
Если отсутствует стандартный образец, то пользуются сборниками спектров (атласами), в которых приводятся спектры веществ и точные условия приготовления пробы для анализа. К настоящему времени изучены и сведены в соответствующие таблицы и атласы инфракрасные спектры более 20 ООО различных соединений, что значительно облегчает практическое использование этого метода.
Метод отличается универсальностью, избирательностью и весьма характерен. В иностранной литературе его часто называют finger print, т.е. отпечатки пальцев, что означает неповторимость инфракрасного спектра каждого соединения.
Надежная идентификация токсических веществ с помощью этого метода может быть проведена только после тщательной очистки извлечений из объекта.
Спектрофотометрия в УФ-области спектра
Спектрофотометрия — метод анализа, основанный на измерении спектров поглощения в оптической области электромагнитного излучения, обусловленного электронными переходами: σ-σ*, η-σ*, π-π*, η-π* (переходы перечислены в порядке уменьшения энергии, необходимой для их осуществления).
Различные электронные переходы в молекулах веществ требуют неодинаковой энергии, а поэтому полосы поглощения располагаются при разных длинах волн.
Наибольшей энергии требует σ-σ* переход. Он связан с возбуждением внутренних электронов и соответствует поглощению в дальней УФ-области (А
Эти переходы не регистрируются в рабочем диапазоне спектрофотометра.
Переход η-σ* связан с меньшими затратами энергии и характерен для органических соединений, содержащих п-электроны, локализованные на орбиталях атомов О, N, Hal, S. Они имеют полосы поглощения при длине волны более 200 нм.
Полосы, соответствующие переходам η-π* и π-π*, характерны для гетероциклических соединений с сопряженными связями, проявляются в области ~ 250-300 нм и имеют большую интенсивность. К этой группе можно отнести соединения, содержащие бензольное кольцо. Это поглощение характерно для многих лекарственных веществ, которые в молекуле имеют фенильный радикал, не связанный с ауксохромными группами. Такие вещества трудно различить по характеру спектра поглощения. Например:
В растворе 0,1 М серной кислоты для них характерны три максимума светопоглоще- ния: при длинах волн 250-252 нм, 256-258 нм, 262-264 нм.
Некоторые органические соединения содержат хромофорные группы, которые связаны с какой-либо ауксохромной группой. При введении в молекулу различных заместителей или при изменении условий измерения (pH среды, природы растворителя) происходит сдвиг полосы поглощения. Если полоса поглощения смещается в сторону более длинных волн, говорят о батохромном смещении. Если полоса сдвигается в сторону более коротких волн — о гипсохромном смещении.
Например:
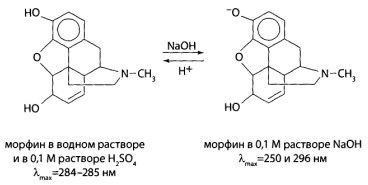
Использование УФ-спектрофотометрии в химико-токсикологическом анализе осложняется присутствием в извлечении примесей эндогенных соединений, что особенно проявляется при анализе трупного материала, подвергшегося гнилостным изменениям. Эндогенные соединения создают так называемое «фоновое поглощение», искажают характер спектра поглощения исследуемого соединения.
Для устранения влияния фонового поглощения в различных методиках используют индивидуальные способы очистки, как на первом этапе изолирования (при настаивании объекта с полярным растворителем), так и на втором этапе (после экстракции токсических веществ органическим растворителем из водной фазы). Если фоновое поглощение остается достаточно высоким, то по характеру спектра не всегда удается идентифицировать вещество.
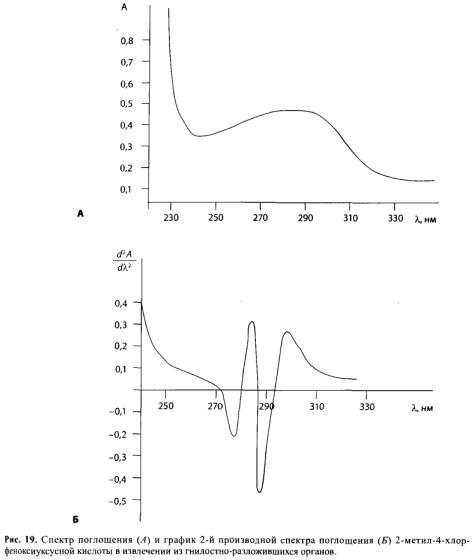
На кафедре токсикологической химии Пятигорской государственной фармацевтической академии разработана методика, позволяющая значительно уменьшить это влияние при определении производных арилоксиалканкарбоновых кислот и некоторых алкалоидов в биологических объектах.
С этой целью регистрируют спектр поглощения извлечения из объекта в области длин волн 200-380 нм и рассчитывают его вторую производную, что позволяет исключить влияние линейного фона на результаты анализа. На графике производной спекгра поглощения исследуемого объекта удается четко обнаружить минимумы, которые соответствуют максимумам поглощения исследуемого соединения. Установлено, что предложенный способ устранения влияния «фонового поглощения» применим при анализе; объектов, подвергшихся гнилостному разложению.
В качестве примера приведен случай обнаружения гербицида 2-метил-хлорфеноксиуксусной кислоты в гнилостно-разложившихся органах. После извлечения яда и очистки вытяжки спектр поглощения не имел характерных полос поглощенш с максимумами при 278 и 289 нм (см. рис. 19А).
На второй производной спектра минимумы при 278 и 289 нм соответствуют максимумам, характерным для указанного гербицида (см. рис. 19Б).
7.2.3. Хроматомасс-спектрометрия
Масс-спектроскопия, масс-спектральный анализ — метод анализа веществ путем определения массы (чаще отношения массы к заряду m/z) и относительного количества ионов, получаемых при ионизации исследуемых веществ или уже присутствующих в изучаемой смеси. Совокупность значений m/z и относительных величин токов этих ионов пред ставляет собой график, который называют масс-спектром вещества.
С помощью масс-спектрометрии можно измерить точную молекулярную массу органического соединения, рассчитать элементный состав, установить химическое и пространственное строение, изотопный состав, провести качественный и количественный, анализ сложной смеси.
При ионизации органической молекулы образуется ион, в котором далее происходят процессы гетеро- и гемолитического разрыва связей с образованием осколочных ионов, которые также подвергаются дальнейшему распаду. Направление распада — важная характеристика каждого класса соединений. Совокупность всех направлений распада составляет характерную для каждого органического соединения схему фрагментации. Если масс-спектр прост, схема фрагментации сводится к одному пути распада. Например, при распаде иона СН3ОН+ последовательно образуются ионы СН2=ОH+ и Н-С=O+. В случае сложных масс-спектров схема фрагментации отвечает многим, часто перекрывающимся: направлениям распада.
Полученный масс-спектр сравнивают со спектром из каталога. Это быстрый, простой способ структурного анализа и идентификации веществ. Он используется для определения загрязнений окружающей среды, контроля продуктов питания, при изучении процессов метаболизма, в криминалистике, а также при анализе биологических объектов на неизвестное ядовитое соединение.
В настоящее время используется сочетание хроматографического и масс- спектрометрического методов. Этот метод получил название хроматомасс-спектрометрии. С помощью хроматографии происходит разделение смеси на отдельные компоненты c помощью масс-спектрометрии проводят обнаружение и количественное определение разделенных веществ смеси.
Хроматомасс-спектрометры выпускаются в двух вариантах — в комбинации с газовым или газожидкостным хроматографом (соответственно ГХ или ГЖХ) для анализа веществ, находящихся в газовой фазе или в комбинации с высокоэффективным жидкостным хроматографом для анализа труднолетучих, полярных и термолабильных веществ.
Если в лаборатории имеется только комплект ГХ — масс-спектрометр, то при анализе малолетучих (барбитураты и др.), полярных соединений и их метаболитов (опиаты и др.) для использования метода необходима дериватизация исследуемых веществ, Дериватизация позволяет исключить потери веществ из-за низкой летучести или сильной полярности. При анализе полярных соединений за счет дериватизации вещества превращаются в менее полярные и более летучие.
Для дериватизации используют реакции различных типов, в основном это:
— ацетилирование: R-O-H → R-O-CO-CH3;
— ацилирование: R-OH → R-O-CO-C2F5;
— силилирование: R-OH → R—О—Si(CH3)3.
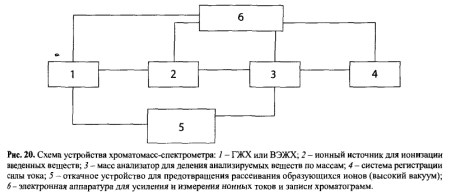
Дериватизацию проводят в герметически закрытых сосудах, снабженных завинчивающимися крышками, или в сосудах с пробками, которые герметично запечатывают с помощью алюминиевых колпачков. В этих сосудах экстракт из мочи упаривают или в них переносят сухой остаток с помощью небольшого объема органического растворителя, который также упаривают досуха. К сухому остатку, содержащему исследуемые вещества, добавляют реагент, не допуская попадания воды, чтобы исключить гидролиз дериватов. Реакция проходит обычно в течение 5-60 мин при температуре менее 100°С. После охлаждения сосуды вскрывают и 1-2 мкл реакционной смеси вносят в инжектор хроматографа.
Наиболее устойчивыми являются ацетилированные производные (реагент: уксусный ангидрид — пиридин, 1:1). Они сохраняют стабильность в реакционной среде при комнатной температуре 48-72 ч. В результате дериватизации появляется возможность более точного определения площадей пиков, форма которых становится более симметричной, н повышается чувствительность определения.
Хроматомасс-спектрометр представляет собой сложный прибор, включающий ионооптическую, высоковакуумную системы, электронную аппаратуру для усиления и измерения ионных токов, а также хроматографический узел для разделения смеси. Блок-схема хроматомасс-спектрометра показана на рисунке 20.
Исследуемую пробу вводят в хроматограф, где происходит разделение смеси веществ. Поток разделенных веществ с газом-носителем проходит через специальный сепаратор, который отделяет газ-носитель от вещества. Затем исследуемое соединение попадает в ионизационную камеру масс-спектрометра. В этой камере частицы веществ ионизируются и поступают в масс-анализатор, где они делятся по массам. Относительные значения сил токов, возникающих за счет ионов с разной массой, измеряются системой регистрации. Масс-спектр смеси веществ представляет собой ряд последовательно расположенных пиков веществ. Деление веществ по массам в анализаторе осуществляется под действием магнитного поля (чаще однородного). При прохождении пучка ионов через магнитное поле ионный пучок фокусируется на щель приемника. В процессе фокусировки пучок ионов с массой ш отделяется от ионов с массой т+Аш. Этот процесс происходит в плоскости щели приемника на расстояние, которое определяется величиной дисперсии (или разделяющей способности) прибора.
Таким образом, разрешающая сила хроматомасс-спектрометра — это мера его способности разделять два иона с определенной разницей их масс.
Детектор (регистрирующее устройство) в приборе осуществляет регистрацию электрических сигналов в определенной последовательности. В современных приборах цифровая регистрация и обработка информации проводится с помощью ЭВМ. Прибор записывает полный спектр и проводит обработку нескольких десятков, а иногда и сотен масс-спектров с сотней пиков в каждом из них с одновременной расшифровкой самого спектра. Непрерывная запись полного ионного тока в течение всего времени хроматографического разделения представляет собой хроматограмму смеси. В момент появленш; максимумов на хроматограмме масс-спектрометр быстро записывает полный масс-спектр каждого компонента с интервалом в 1-2 с. Масс-спектр является качественной хе рактеристикой разделяемой смеси и позволяет идентифицировать ее компонента (предел обнаружения веществ 10 -12 г/мл).
Идентификация веществ, выделенных из биологического объекта, проводится по времени удерживания и соответствующему каждому пику на хроматограмме масс-спектр с определенной величиной отношения массы иона к его заряду. Например, в процессе анализа гашиша при определении каннабиноидов из масс-спектра были выделены характерные отношения масса/заряд 231, 314 и 299. В результате обнаружены 2 вещества, имеющие время удерживания 7,55 и 8,31 мин. Эти вещества были идентифицированы по атласу известных масс-спектров как каннабидиол и тетрагидроканнабинол соответ 1 ственно (Еремин С.К.)
Хроматомасс-спектрометрия является универсальным методом, позволяющим работать с весьма сложными смесями, содержащими всего 10-10 — 10-14 г/мл определяемого компонента, что в миллиарды раз меньше требуемых количеств вещества для обычных1 масс-спектрометров. Это особенно ценно для определения следовых количеств ядовитых веществ в биологических жидкостях, волосах и в трупном материале.
7.2.4. Люминесцентный метод анализа
Этот метод основан на явлении люминесценции. Наибольшее распространение получил анализ, основанный на люминесценции, возбуждаемой УФ-излучением. Для этой цели применяют ртутно-кварцевую лампу типа ПРК и светофильтры УФС-3 и УФС-4, которые почти полностью поглощают видимый свет, но пропускают ультрафиолетовый.
По характеру люминесцентного свечения различают фосфоресценцию — свечение, продолжающееся более или менее длительное время после отключения источника возбуждения свечения и флуоресценцию — свечение, прекращающееся сразу после удаления источника возбуждения.
Интенсивность флуоресценции зависит от присутствия в растворе посторонних веществ. Некоторые вещества способны гасить ее, и в их присутствии интенсивность флуоресценции падает. Например, присутствие ионов хлора значительно ослабляет флуоресценцию хинина. Некоторые вещества вызывают усиление флуоресценции, например серная кислота. Способность некоторых ядовитых веществ флуоресцировать используется в химико-токсикологическом анализе с целью их обнаружения. Эта реакция отличается высокой чувствительностью.
Если органическое соединение обладает кислотными или основными свойствами, его люминесценция меняется в зависимости от pH среды. Например, в кислой среде в присутствии серной кислоты хинин имеет голубую флуоресценцию, в щелочной среде (рН=9) хинин флуоресцирует фиолетовым цветом. Продукты окисления хинина имеют желто-зеленую флуоресценцию.
Флуоресценция используется для обнаружения пахикарпина. Продукты его окисления флуоресцируют красно-оранжевым цветом. Для секуринина характерна желтокоричневая флуоресценция.
Способность ядовитых веществ флуоресцировать или поглощать УФ-лучи используется при определении в биологических объектах лекарственных и наркотических веществ с помощью хроматографии в тонком слое сорбента. При облучении пластинки УФ- излучателем возможно обнаружение некоторых ядовитых веществ. Флуоресцировать могут и трупные яды, выделяемые из объекта вместе с веществами кислотного и основного характера. Например, при облучении УФ-лучами норгарман дает темно-синюю флуоресценцию, триптамин — желто-зеленую. Поэтому использование люминесцентного анализа сочетается с применением других подтверждающих способов и реакций обнаружения.
Люминесцентный метод широко используется также в иммунохимическом анализе для обнаружения и определения многих лекарственных и наркотических веществ.
7.2.5. Микрокристаллоскопический метод
Этот метод основан на образовании характерных кристаллических осадков при действии небольших количеств реактивов на каплю анализируемого раствора на предметном стекле. Полученный осадок исследуют под микроскопом с увеличением в 60 и более раз. Определяют форму кристаллов, их цвет, размер. Кристаллы характерной формы образуются при медленной кристаллизации из разбавленных растворов. Если концентрация вещества большая или в растворе присутствуют примеси эндогенных соединений, форма кристаллов может искажаться, и идентификация вещества затрудняется. В химикотоксикологическом анализе рекомендуется использовать поляризационный микроскоп и определять кристаллографические и кристаллооптические характеристики кристаллов (углы между гранями, показатель преломления, угол погасания, знак удлинения, плеохроизм и др.) для более надежной идентификации веществ. В ряде случаев для характеристики внешней формы кристаллов в химико-токсикологическом анализе употребляют термины, которые не приняты в кристаллографии. Например: «сростки кристаллов в виде летящих птиц», «кристаллы, напоминающие дубовые листья», «густые сростки кристаллов», «чечевицеобразные кристаллы» и т.д. Микрокристаллоскопический анализ используют обычно как подтверждающий на завершающем этапе исследования, когда исследуемое вещество в объекте уже установлено другими реакциями и методами. По образованию осадков и кристаллов характерной формы подтверждают обнаружение многих токсикологически важных соединений.
Например:
1. Производные барбитуровой кислоты идентифицируют и различают между собой по образованию характерных кристаллов с помощью следующих реакций:
- выделение кислотной формы барбитурата;
- с хлорцинкйодом;
- с меднопиридиновым реактивом (реакция Цвиккера);
- с железойодидной комплексной солью;
- с меднойодидной комплексной солью;
- с реактивом Г.Ф. Лозовой, содержащим растворы йодида калия, этиловый спирт и серную кислоту и др.;
2. Некоторые алкалоиды и вещества основного характера образуют характерные кристаллические осадки с реактивами:
- Драгендорфа;
- с солью Рейнеке NH4[Cr (NH3)2 (SCN)4];
- с пикриновой кислотой;
- с реактивом Бушарда;
- с хлоридом ртути (II);
- ализариновым красным;
- роданидом кобальта и др.;
3. Некоторые «металлические» яды образуют характерной формы кристаллы при проведении подтверждающих реакций:
- перекристаллизации из серной кислоты;
- образования тройной соли гексанитрита калия, свинца и меди;
- с солями цезия и йодида калия;
- с хлоридами золота и рубидия;
- с бруцином и бромидом калия;
- с тетрароданомеркуратом аммония;
- образование оксида мышьяка и др.
7.2.6. Фармакологические (физиологические) пробы
Такие испытания проводятся на некоторые ядовитые вещества, которые при воздействии на организм животных вызывают характерные физиологические реакции. Если в ходе химико-токсикологического анализа в исследуемых объектах обнаружены такие алкалоиды, как атропин, кокаин, вератрин, никотин, стрихнин, то в качестве подтверждающего способа используют фармакологические пробы. С этой целью выделенные из объекта алкалоиды хорошо очищают с помощью соответствующих методов (о них мы говорили ранее) и испытывают на животных.
Атропин, кокаин при введении в глаз кошки вызывают расширение зрачка (рис. 21).
При нанесении очищенного экстракта из объекта, содержащего никотин, вератрин, на спинку лягушки она принимает характерную позу (рис. 22 и 23).
При нанесении на спинку лягушки экстракта из объекта, содержащего стрихнин, появляются тетанические судороги сначала от прикосновения к лапкам, затем при сотрясении или ударе. Наконец лягушка вытягивается и погибает в характерной позе (рис. 24).

Фармакологические испытания чаще всего поручаются соответствующему специалисту — фармакологу. При совпадении результатов фармакологического испытания и химико-токсикологического анализа заключение об обнаружении в объекте атропина, кокаина, никотина, вератрина или стрихнина становится более достоверным.
7.2.7. Фармакогностический анализ
В случае обнаружения в содержимом желудка, рвотных массах частей растений, грибов, семян, кусочков индийской конопли проводится фармакогностический анализ. После хи- мического^анализа биологического объекта или параллельно с ним проводится исследование инородных включений растительного происхождения с использованием приемов и методов фармакогностического анализа. В этом случае химик консультируется со специалистом (фармакогностом) или передает ему изъятые части растений для проведения микроскопического анализа. По характерным признакам (волоскам, железкам, друзам, пылинкам, особым клеткам и др.) делается заключение о принадлежности изъятых из объекта включений к определенным видам растений (рис. 25-27).
В сочетании с данными химического анализа делается заключение, явились ли инородные включения причиной отравления или нет. Фармакогностический анализ особенно важен при определении принадлежности частей растений и кустарно изготовленных из них психотропных и сильнодействующих средств к наркосодержащим.
