6.1. План проведения химико-токсикологического анализа
6.2. Характеристика объектов химико-токсикологического анализа
6.3. Подготовка объектов к изолированию ядовитых веществ
6.4. Изолирование токсических веществ путем экстракции и сорбции
6.4.1. Изолирование лекарственных и наркотических веществ амфифильными растворителями
6.4.2. Изолирование подкисленной водой
6.4.3. Изолирование подщелоченной водой
6.4.4. Изолирование наркотических и одурманивающих веществиз мочи твердофазной экстракцией
6.4.5. Экстракция органическими растворителями
6.4.6. Экстракция водой в сочетании с диализом
6.5. Методы минерализации
6.5.1. Методы «мокрой минерализации»
6.5.2. Метод минерализации для обнаружения ртути в объекте
6.5.3. Методы «сухого озоления»
6.6. Методы изолирования «летучих» ядов
6.6.1. Метод перегонки с водяным паром
6.6.2. Методы «микроперегонки» и микродиффузии
Достоверность результатов химико-токсикологического анализа является в большинстве случаев решающим фактором для определения путей лечения при несмертельных отравлениях или одним из главных доказательств причин отравления при смертельном исходе. Это налагает особую ответственность на химика при организации и проведении исследования различных объектов.
Химико-токсикологический анализ предполагает решение двух больших задач: выделение ядовитых веществ из объекта исследования (изолирование) и определение содержания этих веществ в изолированной фазе. Обе эти задачи базируются на разных разделах химии (неорганической, органической, аналитической, физической) и физики. Однако они в значительной степени опираются на собственные предпосылки токсикологической химии. Так, в токсикологической химии под термином «изолирование» чаще всего объединяются три понятия: разделение, концентрирование и выделение. При изолировании ядовитых веществ проводится отделение токсических веществ от субстрата, повышение их концентрации по сравнению с концентрацией в субстрате с последующим выделением в самостоятельную фазу. В то же время процесс изолирования основывается на таких свойствах, как летучесть, основность, кислотность, растворимость ядовитых веществ, которые используются для их разделения и концентрирования.
Выбор методов изолирования определяется обстоятельствами дела, природой объекта, результатами предварительных испытаний. Если отсутствуют точные указания на наличие того или иного вещества в объектах исследования, то используют общую схему изолирования, позволяющую извлечь вещества, проявляющие свойства оснований или кислот.
Для извлечения определенных групп веществ применяют специальные методы, Выбор метода изолирования и очистки определяется следующими факторами:
1. Конкретной практической задачей, т.е. природой объекта, метрологическими параметрами методики.
2. Предысторией объекта (предварительное исследование, обстоятельства отравления, указание на происхождение и т.д.).
3. Сочетаемостью выбранного метода изолирования и очистки с методом последующего обнаружения и определения ядовитого вещества в извлечении.
При разработке методик определения токсических веществ в различных объектах следует учитывать оснащенность лабораторий современными приборами.
6.1. План проведения химико-токсикологического анализа
Необходимость составления плана анализа определяется тем, что объекты исследования нельзя продублировать. При острых отравлениях ядовитыми веществами кровь, моча и другие жидкости организма человека быстро изменяются, и повторный их анализ даст совершенно другие результаты. При смертельных отравлениях вещественные доказательства вторично не могут быть предоставлены.
Составление плана химико-токсикологического анализа зависит от природы и характера объекта исследования, поставленных перед экспертом вопросов, содержания сопроводительных документов и результатов наружного осмотра объекта.
План химико-токсикологического анализа должен быть построен так, чтобы наиболее рационально и с малой затратой времени решить главную задачу — обнаружить и определить количественно ядовитые вещества и (или) их метаболиты в исследуемых объектах.
Составление плана проводится в соответствии с Приказом М3 РФ №289 от 5.10.1989 г. и Приказом М3 РФ №161 от 24.04.2003 г. в следующем порядке.
Осмотр присланного на анализ объекта
Наружный осмотр объекта. Объекты подвергают подробному осмотру и сравнивают с описанием в сопроводительном документе. Обращают особое внимание на особенности упаковки объекта, надписи иа банках, склянках, пакетах, ящиках, коробках, на их содержание, оттиск, целостность печати. Убедившись в полном соответствии, приступают к вскрытию упаковки, что делается осторожно, чтобы предотвратить попадание в сам объект частей печати или упаковки.
Осмотр объекта после вскрытия упаковки. Данные осмотра объекта позволяют предположить, чем произошло отравление, и включить в план анализа в первую очередь исследование на предполагаемые вещества.
Определение природы, характера и запаха объекта. После вскрытия упаковки важно установить, какие органы или их части доставлены на исследование и в каком они состоянии — имеются ли признаки гнилостного разложения. Специфический запах объекта (особенно содержимого желудка) можно уловить при отсутствии резких признаков гниения, так как сероводород и аммиак, образующиеся при этом, могут маскировать запах чужеродных соединений. Характерный запах объекту могут придать многие «летучие» яды. Например, запах горького миндаля указывает на возможное отравление цианидами, запах пиридиновых оснований — на возможное отравление денатурированным спиртом. Можно ощутить характерный запах фенола, сивушного масла, хлороформа, ацетона, формальдегида, этилового спирта и других пахучих веществ.
Определение наличия инородных включений. Объект осматривают вначале визуально, а затем с помощью лупы. Инородные включения могут быть обнаружены в содержимом желудка. Это — части растений, семена, кристаллы солей алкалоидов, металлов, нераспавшиеся таблетки, порошки и др. Все подозрительные инородные включения отбирают при помощи чистого пинцета и анализируют отдельно. Исследование отобранных кусочков растений, грибов, семян, порошка индийской конопли и других включений растительного происхождения производится лицами, имеющими познания в области фармакогнозии.
Определение окраски объекта. Окраска объекта (главным образом содержимого желудка) может также ориентировать на возможное отравление некоторыми ядовитыми веществами.
Например, желтая окраска указывает на возможное отравление хроматами, азотной кислотой, некоторыми анилиновыми красителями, пикриновой кислотой, акрихином. Зеленое, синее или фиолетовое окрашивание встречается при отравлении солями меди и некоторыми красителями, черное окрашивание (с обугливанием) слизистой желудка или одежды может дать указание на наличие концентрированной серной кислоты и т.д. Биологические жидкости (кровь, моча) могут также иметь необычную окраску при отравлении некоторыми ядовитыми веществами. Например, при попадании в организм феиола моча обычно окрашена в оливковый или темио-зеленый цвет за счет продуктов окисления фенола. При отравлении нитробензолом, анилином кровь приобретает шоколадную окраску, при отравлении нитритами, бертолетовой солью — красио-коричневую, при отравлении оксидом углерода — алую.
Предварительные испытания с объектом
Предварительные испытания преследуют цель сократить время исследования объекта, что особенно важно при ненаправленном анализе. Такие испытания позволяют сузить круг веществ в окончательном испытании и определить направление его основного исследования. Обычно для предварительных испытаний подбирают групповые реакции, обладающие высокой чувствительностью. С их помощью можно обнаружить не только токсические, но и терапевтические дозы принятых веществ, а иногда и естественно содержащиеся в объекте соединения. Поэтому делать вывод, что найденное вещество явилось причиной отравления, только по результатам предварительных испытаний недопустимо.
Положительный результат предварительных испытаний указывает на то, что в исследуемом объекте может быть найдено одно вещество или группа веществ, которые дают такие же реакции. В этом случае в план анализа включается основное исследование на эту группу соединений с использованием специальных приемов, методов и подтверждающих реакций.
Отрицательный результат предварительных испытаний указывает на отсутствие соответствующих веществ в исследуемом объекте, и данное вещество (или группа веществ) исключается из плана анализа, после окончания экспертизы делается заключение о его (или их) необнаружении.
Предварительные испытания с содержимым желудка
Определение pH среды проводится в содержимом желудка и имеет значение для предварительного решения вопроса о веществах, которые могли быть причиной отравления. С этой целью применяют индикаторные бумажки, пропитанные фенолфталеином, лакмусом, конго красным и универсальным индикатором. Небольшое количество объекта измельчают, добавляют очищенную воду и взбалтывают. В полученной водной вытяжке определяют реакцию среды путем ее нанесения стеклянной палочкой на полоску индикаторной бумажки.
При наличии минеральных или больших количеств органических кислот синяя лакмусовая бумажка покраснеет, бумажка, смоченная конго красным, посинеет, универсальный индикатор покажет значение рН7. Щелочная среда водных вытяжек может быть обусловлена присутствием в объекте едких щелочей, карбонатов щелочных металлов, аммиака и других соединений. Чтобы установить природу щелочи, к водной вытяжке добавляют 1-2 капли спиртового раствора фенолфталеина — образуется розовое или красное окрашивание. К окрашенному раствору добавляют раствор хлорида бария:
2NaOH + ВаСl2 → 2NaCl + Ва (ОН)2
2NH4OH + BaCl2 → 2NH4Cl + Ва (ОН)2
Na2CO3 + BaCl2 → 2NaCl+ BaCO3
Если в растворе была едкая щелочь или аммиак, то образуется гидроксид бария, гидроксильные ионы в растворе сохраняются, и окраска фенолфталеина ие исчезает. Если в растворе был карбонат натрия, то при добавлении хлорида бария образуется осадок карбоната бария, и окраска фенолфталеина исчезает. Раствор становится бесцветным.
Чтобы решить вопрос о наличии аммиака и его происхождении, часть объекта помещают в колбу, закрывают пробкой, к нижней поверхности которой прикрепляют три индикаторные бумажки: смоченную водой красную лакмусовую, смоченную щелочным раствором ацетата свинца, смоченную щелочным раствором сульфата меди.
Колбу нагревают. Если в объекте присутствует аммиак, красная лакмусовая бумажка меняет цвет на синий, «медная» принимает также синее окрашивание, а «свинцовая» остается без изменения. Если в объекте начались процессы гниения, то образовались эндогенные сероводород и аммиак. В этом случае изменяют цвет все три бумажки: красная лакмусовая и «медная» — на синий цвет, «свинцовая» — на черный. При таком результате делают вывод о невозможности обнаружения аммиака в данном объекте.
CuSO4 + 2NaOH →Cu (OH)2 + Na2SO4
Cu (OH)2 + 4NH3 → [Cu (NH3)4]2+ + 2OH—
Pb (CH3COO)2 + H2S →PbS + 2CH3COOH
Таким образом, определение pH исследуемого объекта дает возможность включить в план химико-токсикологического анализа исследование иа минеральные кислоты (щелочи) или исключить их из этого плана.
Предварительные испытания мочи на некоторые токсические вещества
С мочой из организма человека выделяются чужеродные соединения, как в виде метаболитов, так и в неизмененном состоянии. В последние годы участились случаи отравления различными лекарственными препаратами, наркотическими средствами, алкоголем и его суррогатами. Чтобы оказать срочную медицинскую помощь пострадавшему, требуются быстро выполнимые методы анализа, к числу которых относятся предварительные испытания, проводимые непосредственно с биологическими жидкостями (преимущественно с мочой).
Испытание на этиловый и метиловый спирты. К моче добавляют дихромат калия и сериую кислоту. При наличии спиртов постепенно появляется зеленое окрашивание. Спирты окисляются до соответствующих альдегидов, a xpoм (VI) восстанавливается до хрома (III), соли которого окрашены в зеленый цвет.

Реакция неспецифична.
Испытание на алкилгалогениды. К моче добавляют гидроксид натрия и пиридин. При нагревании на водяной баие появляется розовое или красное окрашивание.
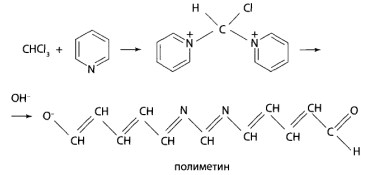
В сильно щелочной среде при нагревании хлороформ вызывает расщепление пиридинового кольца с образованием аниона полиметина (реакция Фудживара). К моче добавляют гидроксид натрия и раствор резорцина. При нагревании появляется розовое окрашивание.
Испытание на ацетон. К моче добавляют растворы гидроксида и нитропруссида натрия — появляется красное или красио-оранжевое окрашивание, которое при добавлении уксусной кислоты переходит в вишнево-красиое.
Испытание на барбитураты. Мочу подкисляют серной кислотой и экстрагируют этиловым эфиром, который отделяют от водной фазы и испаряют досуха. Остаток растворяют в хлороформе и наносят на фильтровальную бумагу, которую после высушивания обрабатывают раствором ацетата кобальта и окуривают парами аммиака — наблюдают фиолетовое окрашивание.
Испытание на производные фенотиазина. К моче добавляют реактив FPN (смесь хлорида железа (III), азотной и хлорной кислот) или серной кислоты и хлорида железа (III) — появляется характерное окрашивание: при наличии аминазина, дипразина — розовое или малиновое, при наличии тизерцииа — фиолетовое, при наличии сонапакса — зеленое, переходящее в синее.
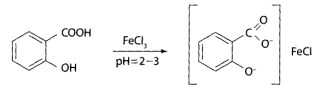
Испытание на салициловую кислоту. При добавлении к моче хлорида железа (III) образуется сине-фиолетовое окрашивание.
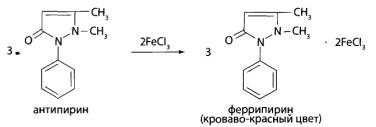
Испытание на производные пиразола. При добавлении к моче хлорида железа (III) наблюдается характерное окрашивание: при наличии антипирина — кроваво-красное, при наличии анальгина или пропифеназона — красно-коричиевое.
Испытание на хинин. К моче добавляют концентрированную серную кислоту и смесь рассматривают в УФ-свете (облучатель, пропускающий свет с длиной волны 254 им) — наблюдают голубую флуоресценцию.
Испытание на хлордиазепоксид. Экстрагируют хлордиазепоксид из мочи хлороформом при значении рН=9-10, хлороформ испаряют, гидролизуют остаток при нагревании в течение часа с 6 М раствором хлороводородной кислоты, а затем проводят реакцию образования азокрасителя.

Испытание на амфетамин. Экстрагируют амфетамин из мочи хлороформом при значении рН=10, упаривают экстракт и наносят остаток иа фильтровальную бумагу. При добавлении реактива Марки появляется оранжевое окрашивание, переходящее в коричневое.
При получении положительного результата предварительного испытания с объектом обнаруженное вещество включается в план основного исследования.
При получении отрицательного результата предварительного испытания с объектом на определенное вещество (или группу веществ) основное исследование не проводится, и делается заключение о необнаружении этого вещества или группы веществ в объекте.
6.2. Характеристика объектов химико-токсикологического анализа
Характерной особенностью химико-токсикологического анализа, как было отмечено ранее, является большое разнообразие объектов, которые могут стать предметом исследования. Основными и наиболее часто встречающимися объектами являются печень, почки, мышечная ткань, кровь, моча, слюна, волосы, ногти. Знание состава исследуемого объекта, поведения и превращения ядовитого вещества в нем имеет особое значение при выборе способа пробоподготовки и метода изолирования чужеродного соединения в случае отравления им.
Внутренние органы (печень, почки, мышечная ткань). Химический состав мышечной ткани, печени, почек сложен и в целом включает следующие вещества: воду — до 70-75%, белки — 18-22%, липиды — 2-3%, неорганические соли — 1-1,5%, углеводы — 2-3%, азогистые экстрактивные вещества — 1-1.7%, безазотистые экстрактивные вещества — 1-1,5%, а также ферменты, витамины.
Благодаря большой молекулярной массе, белки в органах находятся в коллоидном состоянии. Они имеют свободные карбоксильные и аминные группы и являются амфотерными соединениями. При значении рН>7 белок проявляет свойства кислоты, а при значении рН
Наибольшее число белков в тканях представлено альбуминами и глобулинами Изоэлектрическая точка у альбуминов наблюдается при значении рН=4,8, у глобулинов — при значении рН=5,4. Альбумины делятся на растворимые в воде и нерастворимые в насыщенных растворах сернокислого аммония. Глобулины нерастворимы в воде, но растворимы в ней в присутствии различных солей. С глобулинами в организме образуют прочные связи «металлические» яды.
Так как белки выше их изоэлектрической точки имеют отрицательный заряд, они могут связывать многие основания (в том числе алкалоиды и другие органические основания). Чем сильнее основание, тем прочнее оно связывается с белками.
Содержание углеводов в живом организме достигает 3% сухой массы. Основная масса углеводов находится в печени и мышцах в виде полисахарида гликогена.
Липиды (жиры и жироподобные вещества) нерастворимы в воде, растворимы в органических растворителях (эфире, хлороформе, спирте, ацетоне, толуоле). Жиры в основном являются триглицеридами, их содержание может достигать 50%. Кроме того, в организме содержится большое количество фосфолипидов, входящих в состав всех клеток.
Кровь. Количество крови в организме человека составляет 6-7,5% массы тела, у взрослого человека это 5-6 л, у новорожденного ребенка — примерно 240 мл. Кровь состоит из плазмы и взвешенных в ней форменных элементов: эритроцитов, лейкоцитов, тромбоцитов. Преобладают в крови эритроциты. Плотность крови человека — 1,050-1,060. Содержание воды — 75-85%. Кровь имеет слабощелочную реакцию среды (рН=7,35-7,4).
Плазма крови (белковая жидкость после удаления форменных элементов) составляет 4% от массы тела. Плазма на 90% состоит из воды, в которой содержание белков достигает 70-80 мг/мл, в их числе 40-50 мг/мл составляют альбумины. Основная часть других белков (до 25 мг/мл) представлена глобулинами, к числу которых относится фибриноген (примерно 3 мг/мл), отвечающий за свертывание крови.
В плазме содержатся 7,5 мг/мл минеральных солей, 4-8 мг/мл липидов, в том числе 2-2,5 мг/мл фосфолипидов. В плазме (сыворотке) находится много эндогенных низкомолекулярных органических веществ. Среди них гормоны, биогенные амины, витамины, креатин, креатинин, билирубин, мочевая кислота, мочевина и др. Эти соединения могут потенциально мешать проведению химико-токсикологического анализа на ядовитые и наркотические вещества. Многие токсические вещества связываются в крови с белками (иногда до 90%), в основном с альбуминами, реже с глобулинами.
Моча — прозрачная жидкость, окрашенная в желтый цвет (за счет пигмента урохрома) или в оранжевый цвет (за счет пигмента уробилина). Объем мочи в среднем составляет ежесуточно 1,5 л у мужчин и 1,2 л у женщин, что зависит от питьевого режима, и может даже достигать 2-3 л. При стоянии в моче появляется муть, представляющая собой фосфаты, карбонаты, оксалаты, сульфаты металлов, в основном второй группы. Моча содержит те вещества, которые поступают к почкам с кровью, но по составу значительно отличается от состава плазмы. Моча в норме не содержит белок и глюкозу. В моче в 50- 100 раз больше, чем в крови, мочевины, вдвое выше концентрация минеральных солей, в 8-10 раз больше уровень мочевой кислоты, креатинииа. За сутки с мочой выделяется 50-70 г сухих веществ. Эти растворенные вещества повышают плотность мочи в среднем до 1,015-1,025 г/мл. В моче много минеральных веществ — за сутки выделяется примерно 20 г солей (в основном хлориды натрия и калия), что снижает температуру замерзания мочи до -1,3-2,3°С, что следует учитывать при лиофилизации проб. Значение pH среды в моче в норме от 4,5 до 8,0 и зависит от характера пищи. Из эндогенных соединений в моче присутствуют низкомолекулярные продукты метаболизма аминокислот, сахаров, стероиды и др. В моче содержатся вещества, являющиеся продуктами обезвреживания ядов в организме. К ним относятся эфирсерные кислоты, парные соединения с глюкуроновой кислотой, оксигиппуровая кислота и др. В виде парных соединений с глюкуроновой кислотой выделяются с мочой ядовитые вещества, образующиеся при гниении в кишечнике (феиол, крезол, иидол, скатол), многие лекарственные, наркотические вещества и их метаболиты (морфин, эфедрин, фенилалкиламины, барбитураты, производные фенотиазина и др.).
Слюна — продукт секреции желез ротовой полости. Оиа содержит ферменты а-амилазу, мальтазу, иоиы калия, кальция, гидрокарбонаты, белковые соединения (альбумины, липопротеиды и др.). Значение pH слюиы 6,8-7,1. Чужеродные соединения, особенно лекарственные и наркотические вещества, могут связываться с белками слюны и подвергаться ферментативному воздействию. Установлено, что неионизироваиные формы токсических веществ, находящиеся в плазме крови, пассивно диффундируют в слюну и в таких случаях существует прямая зависимость между концентрацией анализируемого вещества в слюне и его концентрацией в плазме крови.
Волосы являются производными эпидермиса. В волосе различают стержень, выступающий иад поверхностью кожи, а также корень, располагающийся в толще кожи и оканчивающийся утолщением (луковицей). Корень волоса (как и стержень) имеет сердцевину, корковый слой и кутикулу. Клетки сердцевины имеют вид тонких пластинок с ядрами и зернами кератогиалииа. Кутикула в верхней части кория состоит из плоских безъядерных чешуек, книзу в них имеются цилиндрические клетки с ядрами. Обычно волосы головы вырастают иа 0,1-0,5 мм в сутки. За месяц они могут удлиниться на 3-15 мм.
Волосы представляют собой сложную комплексную структуру, состоящую, главным образом, из белков, липидов и меланина. Основной субстанцией волос является твердый кератин, отличающийся большой прочностью. Он плохо растворим в воде, устойчив к воздействию химических веществ — кислот, щелочей. Кератин — белковое вещество. Он богат серой (около 4-5%) и аминокислотами (цистеин — около 14%, лейцин — 14%, глютаминовая кислота — 12%, тирозин — 3%), которые способны связывать некоторые металлы.
Мелании придает природную окраску волосу. Ои состоит из полимеров индольно- хинолиновой структуры и способен связываться с большинством физиологически активных веществ.
Липиды волос имеют в своем составе полярные группы, в число которых входят ненасыщенные связи, гидроксильные и эфирные группы, которые образуют связи с лекарственными и наркотическими веществами по различным механизмам.
Волосы представляют собой легкодоступный для отбора и хранения биологический субстрат при химико-токсикологическом анализе как на неорганические, так и на органические яды. Важно то, что наркотические и лекарственные вещества не метаболизи- руются в волосах.
В последние годы установлено, что в волосах наркоманов обнаруживаются опиаты, амфетамины, кокаии, каннабиноиды, фенциклидин, метаквалон и другие яды. Возникает возможность обнаружения лекарственных, наркотических, психотропных средств, сильнодействующих и одурманивающих веществ, некоторых «металлических» ядов в волосах в отдаленные сроки после окончания их приема и даже в тех случаях, когда анализ биологических объектов дает отрицательный результат. В литературе описаны случаи обнаружения опиатов в волосах ирландского поэта Джона Китса спустя 167 лет после смерти. Известно обнаружение морфина в волосах египетской мумии через несколько тысяч лет после гибели организма. Бензоилэкгонин был обнаружен в мумии человека, жевавшего листья кока -2000 лет назад.
Анаболический стероид стаиозолол в 1974 г. был запрещен для использования спортсменами. Этот препарат после 4-18 иед. применения и прекращения приема за месяц до начала соревнований не обнаруживался в моче, но в волосах его можно было обнаружить спустя год и более после приема.
Ногти. Подобно волосам, ногти также являются производными эпидермиса. Это роговые пластинки, располагающиеся на тыльной поверхности концевых фаланг пальцев рук и ног. Ногтевая пластинка образована плотно прилегающими друг к другу роговыми чешуйками плоской формы, заполненными твердым кератином, устойчивым к воздействию химических веществ. Белок кератин содержит цистин, аргинин, тирозин, лизин, фенилаланин, триптофан, гистидин и др. Ногти содержат 10,1-13,7% воды, 0,15-0,76% жироподобных веществ (холестерин и его эфиры), минеральные вещества (кальций, фосфор, цинк, мышьяк и др.). Установлено, что в ногтях способны накапливаться наркотические вещества и прежде всего — опиаты.
6.3. Подготовка объектов к изолированию ядовитых веществ
Внутренние органы (печень, почки, мышечная ткань). Органы измельчают до размеров кусочков 0,5×0,5×0,5 см. Естественно, что более мелкое измельчение приводит к увеличению экстракционной поверхности и одновременно к значительному увеличению количества балластных эндогенных соединений (белков, ферментов, продуктов распада белковых молекул, пигментов и др.) в извлечении. При этом химик вынужден будет применить особые способы очистки, что приведет к потере токсических веществ, особенно при их следовых количествах в объекте.
Рекомендуется использовать также вымораживание объекта при температуре -30-40°С. При этом в органах образуются льдинки, которые разрывают клетки, ткаии и способствуют выходу токсических веществ в окружающую среду, что увеличивает процент изолирования искомых соединений.
Можно использовать лиофтизацию, т.е. высушивание объекта при низких температурах в вакууме. Это приводит к потере объектом воды и получению при изолировании более чистых извлечений.
Если объект законсервирован спиртом, его осторожно удаляют. В случае подозрения на отравление летучими соединениями мышьяка, ртути объект подщелачивают перед удалением спирта карбонатом натрия.
Для анализа обычно берут иавеску объекта 5,25, 50 или 100 г (зависит от конкретной методики).
Кровь. Ядовитые вещества и их метаболиты в крови находятся в свободном виде или могут быть связаны с белками (альбуминами, глобулинами). При пробоподготовке крови к экстракции используют приемы, позволяющие разрушить комплекс анализируемого вещества с белком. Для этой цели рекомендованы следующие методы.
Добавление к крови смешивающихся с водой органических растворителей (этилового, метилового спиртов, ацетонитрила, ацетона). Их количество в 10 раз превышает объем крови. Эффективность очистки от белков зависит от величины диэлектрической проницаемости (е) используемого растворителя. Для ацетона е=31, для этилового спирта — 26,8, для метилового спирта -23,1, для воды — 80. При понижении диэлектрической проницаемости сила притяжения между молекулами растворенных веществ возрастает. В результате под влиянием ацетона и спиртов (этилового и метилового) происходит агрегация молекул белковых веществ крови, понижается растворимость и происходит выпадение их в осадок.
Добавление химических агентов для коагуляции белков, кислот (трихлоруксусиой, хлорной), солей тяжелых металлов (например, солей бария).
Термическая обработка крови. Этот метод используют для ядовитых веществ, которые не разлагаются при повышенной температуре. Ои не рекомендуется при анализе объекта, содержащего термолабильиые соединения.
Процесс пробоподготовки крови к анализу — весьма ответственная операция, при которой возникает опасность адсорбции значительного количества анализируемого вещества на скоагулироваином белке.
Моча. С мочой токсические вещества выделяются как в неизмененном состоянии, так и в виде метаболитов и конъюгатов с еериой, уксусной, глюкуроновой кислотами. Пробоподготовка мочи к экстракции токсических веществ и их метаболитов включает проведение разрушения конъюгатов с указанными кислотами. Для этой цели проводят неспецифический кислотный или специфический ферментативный гидролиз.
Кислотный гидролиз является более быстрым и простым в осуществлении. Однако вследствие неспецифичности реакции расщепления ковалентной связи и жестких условий проведения в среде концентрированной кислоты при кипячении в течение длительного времени или при нагревании в автоклаве под давлением, образуется большое количество побочных продуктов. Экстракция органическим растворителем из полученного гидролизата дает высокий фои и много посторонних пиков при проведении анализа методом газовой хроматографии. Кислотный гидролиз проводится в герметично закрытых сосудах, которые помещают в водяные бани или специальные нагревательные блоки. Можно использовать кипячение с обратным холодильником или нагревание при 100- 125°С в автоклаве под давлением 12-15 пси.
Всемирной Организацией Здравоохранения (ВОЗ) для опиатов рекомендуется проводить кислотный гидролиз по следующей методике: в пробирку емкостью 50 мл вносят 10 мл мочи, добавляют 1 мл концентрированной хлороводородной кислоты, герметично закрывают пробирку и нагревают в водяной бане при 100°С около 60 мии. После охлаждения проводят экстракцию органическим растворителем.
Ферментативный гидролиз проводится в присутствии одного или смеси ферментов β-сульфатазы и β-глюкуронидазы в мягких условиях, что уменьшает образование побочных продуктов, и гидролизат получается более чистым. Существенным недостатком метода являются необходимость соблюдения строгих условий гидролиза: pH, температуры, состава буфера, активности ферментов, — а также длительность процесса (12-20 ч). Легче гидролизуются конъюгаты, образованные через кислород фенольного гидроксила, более длительно — конъюгаты, связанные через кислород спиртовой (гидроксильной) группы.
Методика, рекомендованная ВОЗ для ферментативного гидролиза опиатов, сводится к следующему: в 5-10 мл мочи создают рН=7 с помощью уксусной кислоты, затем добавляют 0,1 мл 0,1 М ацетатного буферного раствора с рН=5,5 и 0,02 мл фермента (75 ЕД/мл) на каждый мл мочи. Смесь оставляют на 24 ч при 37°С или иа 1 ч при 55°С. Температура не должна превышать 55°С, иначе фермент может подвергнуться денатурации. После охлаждения проводят экстракцию органическим растворителем.
Слюна. Для снижения активности ферментов слюну при хранении замораживают. Перед экстракцией ее разбавляют водой очищенной в отношении 1:3, после чего экстрагируют токсические вещества органическими растворителями при соответствующем значении pH.
Волосы, ногти. Для удаления внешних загрязнений волосы и ногти отмывают 2 М раствором хлороводородной кислоты и метанолом (или этанолом), затем сушат при комнатной температуре и отбирают для анализа навеску 30—40 мг.
6.4. Изолирование токсических веществ путем экстракции и сорбции
Наиболее распространенный способ выделения ядов — их извлечение из объектов с помощью различных растворителей. В токсикологической химии путем экстракции выделяют ядовитые вещества из твердой фазы, различные ткани, органы, растительные объекты, и из жидкой фазы: кровь, моча, слюна, промывные воды желудка, перитонеальная жидкость и др. Выделение токсических веществ из жидкой фазы (реэкстракция) часто используется и для очистки извлечений от примеси эндогенных соединений. Таким образом, жидкостная экстракция является одним из главных способов выделения многих ядов из биологических объектов. С помощью жидкостной экстракции рекомендуется проводить выделение из объектов различных групп соединений. Среди них лекарственные, наркотические вещества, пестициды и др.
Лекарственные и наркотические вещества
В токсикологической химии рассматриваются следующие группы лекарственных и наркотических веществ, производные:
— пиридина и пиперидина (никотин, анабазии, пахикарпин);
— тропана (атропин, кокаин, скополамин);
— хинолина (хинии);
— тетрагидроизохинолина (наркотин);
— бензилизохинолина (папаверин);
— фенантренизохинолина (опиаты: морфин, кодеин, наркотии, тебаин, этилморфин, диацетилморфин, орипавин);
— опиоидные анальгетики — промедол, фентанил, трамал, метадон;
— индола (стрихнин, бруцин, LSD и др.);
— пурина (кофеин);
— фенциклидин и его аналоги (тиеиоциклидин, ролициклин, этициклидин);
— производные барбитуровой кислоты: фенобарбитал, барбамил, бутобарбитал, этамииал-натрий, барбитал;
— производные 1,4-бензодиазепина: хлордиазепоксид, диазепам, оксазепам, нитразепам;
— производные п-аминобензойной кислоты: новокаин, иовокаинамид;
— производные фенотиазина: аминазин, дипразин, левомепрамазин, тиоридазин;
— каннабиноиды: каннабидиол, каннабинол, тетрагидроканнабинол, тетрагидрокан- набиноловая кислота;
— фенилалкиламины: эфедрин, эфедрон, амфетамин, метамфетамин.
Характерной особенностью приведенных веществ является их способность давать ионизированные и неионизированные формы. Существование различных форм зависит от величины константы ионизации вещества и значения pH среды. Для кислот константа ионизации определяется уравнением:
Ка = ([Н+] * [А—])/[НА]
или в логарифмической форме: рКа = pH + lg [НА] — lg [А].
Для оснований:
Ка = ([Н+] * [В])/[НВ+]
в логарифмической форме: рКа = pH + lg [В] — lg [ВН+].
Исходя из этих уравнений, можно считать, что кислота будет полностью (99,9%) ионизирована при рН=рКа+3, а основание — при рН=рКа-3. Эти закономерности справедливы для водных растворов, В смешанных или неводных растворителях эти соотношения могут изменяться, иногда на значительную величину.
Достичь условий полной ионизации веществ удается не всегда. В этих случаях ограничивают значения pH до величины рКа±2 и даже меньше. При этом следует учитывать, что при рН=рКа только 50% вещества будет находиться в ионизированной форме. В таблице 3 представлены константы диссоциации некоторых токсикологически важных соединений.
Эти закономерности используют как при извлечении ядовитых веществ, так и при очистке полученных экстрактов. Соли кислот и оснований хорошо растворимы в воде и спирте и практически не растворяются в органических растворителях. Свободные кислоты и основания хорошо растворимы в органических растворителях и плохо растворимы в воде и спиртоводных смесях.
Метод извлечения ядовитых веществ из твердых объектов (твердая фаза) водным или спиртовым раствором кислоты или основания практически всегда является первым этапом изолирования. На этом этапе измельченные внутренние органы заливают экстрагентом, устанавливают необходимое значение pH и настаивают в течение определенного времени. Выбор растворителя и время настаивания зависят от многих факторов.
Вода и спирт как наиболее часто применяемые растворители отвечают многим требованиям, предъявляемым к экстрагентам. В них хорошо растворяются многие соли оснований и кислот.
Время настаивания зависит от скорости проникновения экстрагента внутрь ткани. Продолжительность настаивания определяется моментом наступления равновесия между концентрацией токсического вещества в ткани и окружающей жидкости.
Вода имеет большее сродство к тканям. Она содержится в большом количестве в органах человека: в мышцах — до 75-78%, в печени — до 70-75%. Поэтому вода легко проникает в ткани. Время настаивания объекта с водой и наступление равновесия составляет 1-2 ч.
Спирт денатурирует белковые молекулы на поверхности кусочков биологического материала, его проникновение в ткани затрудняется, и равновесие возникает не менее чем через 6 часов.
Для создания необходимого значения pH среды объект с экстрагентом подкисляют органической или неорганической кислотой. Природа кислоты может оказывать влияние на степень изолирования алкалоидов и органических оснований из биологического материала. Чем лучше растворяются соли ядовитых веществ в данном растворителе, тем лучше они будут извлекаться.
Применение минеральных кислот для подкисления объектов ограничивается возможностью гидролиза эфироподобных токсических веществ (атропин, кокаин и др.) и белковых молекул. В.Ф.Крамаренко установлено, что такие явления не происходят, если для подкисления взята разбавленная минеральная кислота.
При необходимости одновременного определения ядовитых веществ и их метаболитов могут возникнуть трудности в подборе условий их извлечения. Метаболиты часто значительно отличаюгся по физико-химическим свойствам от самих ядов. Поэтому для их извлечения требуются иные растворители и значение pH среды. В таких случаях изолирование ядов и их метаболитов иногда рекомендуется проводить из разных навесок объекта.
Таблица 3. Показатель константы ионизации (рКа) некоторых веществ кислотного и основного характера
|
Вещество |
рКа1 |
рКа2 |
|
Апоморфин |
5,88 |
|
|
Барбитал |
7,97 |
|
|
Бутобарбитал |
7,98 |
|
|
Барбамил |
7,96 |
|
|
Фенобарбитал |
7,45 |
|
|
Этаминал |
8,20 |
|
|
Наркотин |
7,83 |
|
|
Нитразепам |
2,30 |
11,00 |
|
Стрихнин |
6,00 |
11,70 |
|
Атропин |
10,00 |
|
|
Бруцин |
6,04 |
11,70 |
|
Кодеин |
6,05 |
|
|
Кокаин |
8,70 |
|
|
Кофеин |
0,60 |
|
|
Морфии |
6,17 |
10,00 |
|
Никотин |
6,16 |
10,86 |
|
Папаверин |
8,09 |
|
|
Хинин |
5,97 |
9,88 |
Для изолирования слабых кислот, в частности барбитуратов, используют подщелоченную воду. При этом образуются натриевые соли барбитуратов, которые хорошо извлекаются водой.
Выбор величины pH при изолировании обычно учитывает только возможность извлечения ядовитых веществ. Однако пептиды тканей также могут ионизироваться и переходить в раствор. Оптимальным значением pH, препятствующим такому переходу, было бы значение pH, равное изоэлектрической точке белка. В реальных условиях этого достичь не удается. Поэтому извлечения из биологических объектов, полученные на первом этапе, всегда содержат примеси белковых молекул и других эндогенных веществ, а поэтому требуют применения различных способов очистки.
Очистка вытяжек из биологического материала на первом этапе изолирования
В нейтральной среде белки мало растворимы в воде и спирте. Однако при подкислении они в большей степени растворяются в воде. Значительно сильнее в подкисленной воде способны растворяться пептиды и аминокислоты. При анализе биологического материала, находящегося в состоянии гнилостного разложения, количество извлекаемых эндогенных соединений увеличивается.
По составу примесей водная и спиртовая вытяжки отличаются между собой. В водном извлечении содержится больше низкомолекулярных пептидов и аминокислот, В спиртовой вытяжке содержатся примеси липопротеидов, липидов, нтомаинов, некоторых аминокислот. Белки в спиртовое извлечение переходят в меньшем количестве, чем в водное, так как спирт вызывает денатурацию белка и уменьшает его растворимость.
Очистка вытяжек от балластных веществ достигается несколькими способами: осаждением примесей различными реагентами, фильтрованием, центрифугированием, гель- хроматографией и другими методами.Осаждение белковых молекул методом высаливания проводится путем добавления к водной вытяжке из объекта электролитов. В результате изменяется ионная сила раствора, которая определяется уравнением:
J=1/2ΣCi · Zi2
где J — величина ионной силы; Ci — концентрация электролита; Zi2 — квадрат заряда иона электролита.
При изменении величины ионной силы от 0 до 0,05-0,1 растворимость многих веществ может увеличиваться. При высоких концентрациях электролитов в растворе, особенно при добавлении их до его насыщения, белки выпадают в осадок за счет резкого снижения их растворимости. Поэтому для высаливания белков используют высокие концентрации солей.
В качестве электролитов при очистке извлечений из трупного материала применяют соли одновалентных катионов: сульфат аммония — (NH4)2SО4, вольфрамат натрия — Na2WО4 и др.
Метод высаливания является одним из эффективных способов осаждения примесей из вытяжек, полученных при настаивании гнилостно-разложившегося биологического материала с подкисленной водой.
Осаждение белковых молекул кислотами, спиртом и нагреванием. Осаждение белков из вытяжек можно проводить с помощью трихлоруксусной, вольфрамовой, фосфорновольфрамовой и фосфорномолибденовой кислот.
Белковые вещества могут осаждаться после их денатурации путем нагревания или при добавлении этилового спирта.
Необходимо отметить, что осаждение примесей указанными методами может привести к потере исследуемых соединений, так как не исключено соосаждение ядовитых веществ и их адсорбция на поверхности образующихся осадков белковых молекул. Кроме того, ядовитые вещества, в молекуле которых имеются эфирные группы, при нагревании могут быть разрушены.
Фильтрование и центрифугирование. Эти способы позволяют отделить вытяжки от механических загрязнений и от образовавшихся продуктов осаждения. Фильтрование — процесс более длительный, и кроме того, материал фильтров частично адсорбирует ядовитые вещества.
Центрифугирование является эффективным способом отделения примесей и осажденных белков от основного раствора. Современные центрифуги позволяют достигать до 15 тысяч оборотов в минуту и получать сравнительно чистую надосадочную жидкость.
Гель-хроматография используется для очистки водной вытяжки из биологических объектов от примесей балластных веществ. Этот метод основан на различной способности молекул проникать в поры неионогенного геля, который служит неподвижной фазой. Для гель- хроматографии в токсикологической химии используют гидрофильные полимерные сорбенты, в частности сефадексы. Например, сефадекс G-25. Это декстран с поперечными сшивками, в воде хорошо набухает, образуя гель, стабильный в разбавленных кислотах и щелочах.
При работе гель помещают в колонку диаметром 2,5 см и высотой 43-45 см. Водную вытяжку из объекта пропускают через колонку. Крупные молекулы белков и пептидов не проникают или проникают только в часть пор. Когда колонку промывают элюентом, первыми вымываются наиболее крупные молекулы, последними — низкомолекулярные вещества.
Метод гель-хроматографии рекомендуется в химико-токсикологическом анализе при изолировании барбитуратов из трупного материала. Он позволяет получать достаточно чистые вытяжки.
Экстракция примесей из вытяжек на первом этапе применяется очень редко. Это связано с тем, что трудно подобрать условия, при которых возможно отделить большие количества примесей от исследуемых веществ, содержащихся часто в объекте в малых концентрациях.
Жидкость-жидкостная экстракция
Эффективным и распространенным методом в химико-токсикологическом анализе является жидкость-жидкостная экстракция. Этот метод применяется при извлечении ее- ществ из крови, лимфы, слюны, перитонеальной жидкости, мочи и промывных вод желудка. Он также используется на втором этапе при изолировании ядовитых соединений из трупного материала.
Жидкость-жидкостная экстракция — это метод выделения, разделения и концентрирования веществ, основанный на распределении растворенного вещества между двумя жидкими несмешивающимися фазами. В токсикологической химии чаще всего одной жидкой фазой является вода, другой — органический растворитель. Органический растворитель должен обладать высокой растворяющей способностью для анализируемого вещества, иметь низкую температуру кипения. Однако выбор органических растворителей невелик. Наиболее часто для этой цели используют диэтиловый эфир и хлороформ. Выбор органического растворителя определяется свойствами анализируемого соединения.
В условиях равновесия отношение концентраций вещества в обеих фазах представляется константой распределения (К), которая не зависит от общей концентрации вещества.
К = [Со] / [Св]
где Со и Св — равновесные концентрации вещества в обеих фазах (органической, водной) в одной и той же форме.
Однако при экстракции могут происходить различные процессы: ассоциация, диссоциация, сольватация, комплексообразование. Практическое значение при химикотоксикологическом анализе имеет отношение общих (аналитических) концентраций экстрагируемого вещества, которое называют коэффициентом распределения (D).
D = Со / Св
где Со и Св — общие концентрации вещества в органической и водной фазе (независимо от формы существования вещества).
Коэффициент распределения определяют экспериментально в реальных условиях. Его значение используют для разработки условий экстракции ядовитого вещества.
Степень извлечения (процент экстракции) при однократной экстракции рассчитывают по формуле:
R = (100 · D) / (D+Vв/Vо)
где R — степень экстракции, %, D — коэффициент распределения, Vв и Vо — равновесные объемы водной и органической фаз.
Эта формула позволяет рассчитать объем органической фазы для определенного процента извлечения вещества при однократной экстракции.
Пример. Найти объем органического растворителя для извлечения 99% вещества из 100 мл водного раствора при однократной экстракции. Коэффициент распределения равен 20.
Из предыдущей формулы находим:
Vо = (R · Vв) / ( (100-99) · 20)
Подставляя указанные значения R и D, находим:
Vо = (99 · 100) / ( (100-99) · 20) = 495 мл
При многократной экстракции удается добиться полной экстракции значительно меньшим объемом органического растворителя. В этом случае расчет степени экстракции проводится по формуле:
R = 100 · [1- (Vв/ (D·Vo+Vв))n]
где п — кратность экстракции.
Для приведенного выше примера при D=20, объеме водной фазы 100 мл достаточно использовать трехкратное извлечение органическим растворителем объемом 25 мл.
R = 100 · [1- (100/ (20·25+100))3]
Таким образом, данным количеством растворителя можно полнее извлечь растворенное вещество, если проводить экстракцию многократно малыми порциями растворителя.
По приведенной выше формуле можно найти необходимую кратность экстракции при заданных условиях с целью достижения требуемой степени экстракции. Из вышеприведенных формул можно найти:
n = [lg (1- (R/100))] / [lg (Vв/ (D·Vo+Vв)]
Используя данные взятого примера VB=100 мл; V0=25 мл; D=20, найдем число экстракций для извлечения 99% вещества:
n = [lg (1- (99/100))] / [lg (100/ (500+100)] = -2 / -0,778 = 2,57 (округлено 3)
Добавление электролитов в водную вытяжку оказывает высаливающее действие за счет понижения растворимости ядовитых веществ в воде. В результате повышается степень их экстракции органическим растворителем.
Влияние pH зависит от характера извлекаемого вещества:
— при экстракции органическим растворителем вещество в водной фазе должно находиться в молекулярном, неионизированном состоянии;
— для веществ кислотного характера необходимо создать в растворе рН=рКа-2, а для оснований — рН=рКа+2;
— для веществ нейтрального характера значение pH особого значения не имеет. Эти соединения экстрагируются как из кислой, так и из щелочной среды;
— для веществ, являющихся амфолитами, расчет pH для экстракции производится иначе. В этих соединениях имеются кислотная и основная группы, и для каждой известна величина рКа.
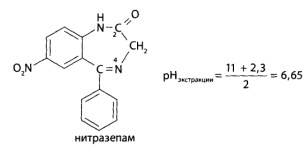
В качестве примеров можно привести расчет оптимальных значений pH для экстракции морфина и нитразепама.

Нитразепам проявляет кислотные свойства за счет имидольной группы в положении 2 (рКа,=11,0) и основные свойства за счет азота в положении 4 (рКа2=2,3).
Для веществ, имеющих в молекуле более одного атома азота, расчет pH для максимальной экстракции проводится с использованием рКа более сильного основания. Например, алкалоид хинин:
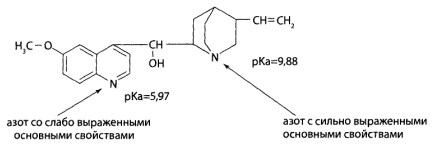
Расчет максимального значения pH для экстракции хинина проводят по рКа азота хинуклидинового кольца:
рНэкстракции = рКа + 2 = 9,8 + 2 = 11,8.
Очистка экстрактов на втором этапе изолирования
При извлечении токсических веществ из водной фазы в органической фазе вместе с токсическими соединениями будут содержаться так называемые соэкстрактивные вещества, среди которых жиры, белки, продукты их распада, птомаины, красящие, дубильные и другие вещества. Эти примеси мешают анализу, загрязняют колонки хроматографов, могут привести к получению ложноположительных результатов. Например, птомаины — продукты распада аминокислот в подвергшемся разложению объекте — дают при анализе характерное сведение в УФ-свете, осадки с общеалкалоидными реактивами осаждения и т.д.
Для очистки экстрактов используют возгонку, реэкстракцию, электрофорез, хроматографию в тонком слое сорбента и др.
Возгонка (сублимация) применима для веществ, способных при нагревании возгоняться без разложения. Это барбитураты, салициловая и бензойная кислоты и некоторые алкалоиды. С этой целью после удаления органического растворителя остаток нагревают. Возгон, содержащий исследуемые вещества, осаждается на охлаждаемой с помощью мокрой ваты или марли поверхности (обычно на стекле). При этом соэкстрактивные соединения не возгоняются. Полученный возгон подвергают анализу.
Электрофорез — разделение веществ под действием внешнего электрического поля. Разделению подвергаются вещества, находящиеся в ионизированном состоянии. С этой целью часто используют лист фильтровальной или специальной бумаги для электрофореза, на который наносят 1/25 часть в виде точки и рядом 1/2 часть в виде полосы полученного из объекта экстракта. Бумагу смачивают буферным раствором с определенным значением pH, помещают в аппарат для горизонтального электрофореза, устанавливают необходимую величину напряжения. Условия подбирают таким образом, чтобы степень миграции (передвижения в электрическом поле) искомых соединений и соэкстрактивных веществ была разной или чтобы разделяемые вещества имели неодинаковый заряд и перемещались к противоположным полюсам. Полученную электрофореграмму высушивают и зону, по которой мигрировали вещества из 1/25 части экстракта, обрабатывают подходящим реагентом-проявителем. Непроявленную часть элекгрофореграммы, соответствующую обнаруженному с помощью реагента-проявителя пятну, вырезают, измельчают, элюируют исследуемое вещество соответствующим растворителем и анализируют.
Реэкстракция основана на переведении веществ из водной фазы в органическую при изменении pH среды. Этот метод применяют для очистки остатков, содержащих барбитураты и некоторые алкалоиды. Используется различная растворимость их молекулярных и ионизированных форм в воде и в органических растворителях. При очистке остатков, содержащих барбитураты, используется их способность к лактам-лактимной (имидо- имидольной) таутомерии. Экстракт, полученный из водной вытяжки при рН=2, содержит барбитураты в лактамной (имидной) форме:
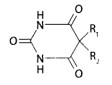
После испарения органического растворителя к остатку добавляют раствор щелочи до рН=10. Барбитураты переходят в соли монолактимной формы. Их особенностью является растворимость в воде и нерастворимость в органическом растворителе.
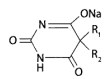
Примеси из полученного щелочного раствора экстрагируют диэтиловым эфиром. Очищенный щелочной раствор подкисляют до рН=2. При этом барбитураты вновь переходят в лактамную форму, способную экстрагироваться органическим растворителем.
Особые трудности представляет анализ гнилостно разложившегося биологического материала, содержащего трупные яды.
Очистка экстрактов от птомаинов. При очистке остатков, содержащих алкалоиды и птомаины, использование реэкстракции основано на способности алкалоидов (оснований) образовывать соли в слабокислой среде, а птомаинов (оснований) — в сильнокислой среде. Экстракт, полученный из водной вытяжки при рН=8-10, содержит алкалоиды и птомаины в виде оснований. После испарения органического растворителя такой остаток окрашен в бурый цвет и маслянистый на вид. Остаток после испарения органического растворителя растворяют в воде и добавляют щавелевую кислоту до слабокислой реакции среды, алкалоиды переходят в соли, а птомаины остаются в виде оснований. После экстракции раствора хлороформом в органическую фазу переходят птомаины-основания, а алкалоиды-соли остаются в растворе, который затем подщелачивают раствором аммиака до рН=8-10 и экстрагируют очищенные алкалоиды-основания хлороформом. Методы реэкстракции часто сочетают с хроматографической очисткой.
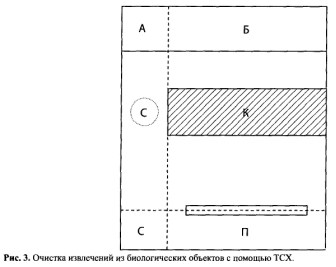
Хроматография в тонком слое сорбента (ТСХ). Очистка извлечений с помощью ТСХ рекомендуется на последнем этапе химико-токсикологического анализа, когда необходимо определить найденное ядовитое вещество количественно или подтвердить его обнаружение с помощью физико-химических методов.
С этой целью определенную часть экстракта наносят на пластинку в виде полосы (II) или в виде капель, а рядом наносят «стандарт» — раствор известного вещества — С (около 10-15 мкг) (рис. 3).
Пластинку помещают в частную систему растворителей, в которой происходит четкое разделение ядовитого вещества, его метаболитов и эндогенных соединений. Когда растворитель поднимется по пластинке на высоту 10 см, ее вынимают, высушивают С помощью стеклянной пластинки закрывают часть Б и обрабатывают реагентом-проявителем ту часть пластинки (А), по которой двигался «стандарт». При этом фиксируют только «пятно-стандарта» С. Затем стеклянную пластинку снимают и зону сорбента (К), соответствующую «пятну-стандарту», соскабливают и элюируют подходящим растворителем (растворами кислот, щелочей, аммиака, спирта и др.).
Полученный элюат используют для обнаружения или количественного определения найденного ядовитого вещества с помощью ГЖХ, ВЭЖХ, УФ-спектрофотометрии, хроматомасс-спектрометрии и др.
6.4.1. Изолирование лекарственных и наркотических веществ амфифильными растворителями
Амфифильные растворители (метанол, этанол, ацетонитрил, ацетон) имеют сродство к гидрофильным и гидрофобным участкам мембран клеток, а поэтому способны легко проникать внутрь клеток и извлекать лекарственные и наркотические вещества. Известно, что в ионизированной форме (при рН=2) лекарственные и наркотические вещества извлекаются полярными растворителями — водой и спиртом, в молекулярной (неионизированной) форме — липофильными (неполярными) растворителями. С помощью амфифильных растворителей лекарственные и наркотические вещества способны извлекаться как в ионизированной, так и в неионизированной форме. При изолировании не обязательно подкислять или подщелачивать объект.
Изолирование подкисленным спиртом
Метод Стаса-Отто. Метод предложен в 1851 г. бельгийским химиком Ж.С.Стасом для изолирования никотина из внутренних органов трупа. В 1856 г. Юлий и Роберт Отто ввели в этот метод очистку от примесей с помощью органических растворителей. В настоящее время он известен как метод Стаса-Отто. На протяжении многих десятилетий этот метод подвергался различным модификациям с учетом факторов, влияющих на изолирование ядовитых веществ. В настоящее время метод применим для выделения не только алкалоидов, но и веществ кислотного, слабоосновного характера, в том числе синтетических лекарственных и наркотических средств.
Схема метода
1-й этап. Навеску объекта массой 100 г настаивают три раза по 24 ч с 96% этиловым спиртом, подкисленным щавелевой кислотой до рН=2,5-3,0. Спиртовые вытяжки упаривают при 40°С и в полученном остатке многократно осаждают белки 96% спиртом. Очищенный остаток растворяют в 20-25 мл воды.
2-й этап. Водный раствор последовательно экстрагируют хлороформом при рН=2 ирН=8-10 после подщелачивания 25% раствором аммиака. В экстракт при рН=2 переходят вещества кислотного и слабоосновного характера. В экстракт при рН=8-10 переходят вещества основного характера.
Оценка метода. Достоинство метода состоит в том, что он позволяет извлекать вещества различной природы. Кроме того, этиловый спирт вызывает осаждение белков, что очень важно при работе с трупным материалом.
Недостатком метода являются длительность выполнения и возможные потери искомых веществ за счет адсорбции их белками при осаждении и фильтровании. Однако несмотря на это и на появление новых методов изолирования, метод Стаса-Отто не теряет значения, особенно при исследовании объектов, подвергшихся сильным гнилостным изменениям.
Метод Е.М. Соломатина. Экстракция подкисленным спиртом хорошо обоснована Е.М.Саломатиным при определении производных фенотиазина в биологических объектах. Обладая слабыми основными свойствами, производные фенотиазина при рН=2-3 образуют ионизированную форму, а при рН>7 — неионизированную.
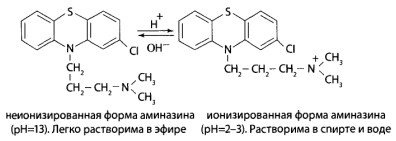
Используя эти свойства, удалось выделить и очистить от примесей анализируемые соединения и их основные метаболиты — сульфоксиды.
Схема метода
1 -й этап. Объект заливают 96% этиловым спиртом, подкисляют щавелевой кислотой до рН=2-3 и настаивают 3 раза по 2 ч. Спиртовые вытяжки упаривают при 40°С до густоты сиропа. В остатке осаждают белки 96% спиртом, фильтруют и выпаривают досуха. Очищенный остаток растворяют в 100 мл воды, нагретой до температуры 40-60°С, охлаждают и фильтруют.
2-й этап. Фильтрат экстрагируют дважды диэтиловым эфиром при рН=2-3, Затем водную фазу подщелачивают 50% раствором гидроксида натрия и вновь экстрагируют диэтиловым эфиром. Объединенные эфирные вытяжки взбалтывают с 0,5 М раствором серной кислоты и полученный кислый водный реэкстракт используют для обнаружения производных фенотиазина.
Оценка метода. По данным автора, метод позволяет достичь максимальной экстракции при изолировании и уменьшить потери исследуемых веществ как при изолировании, так и при экстракции.
Изолирование нейтральным ацетоном
Метод разработан В. А. Карташовым. В качестве экстрагента им предложен амфифильный растворитель ацетон, который способен смешиваться как с водой, гак и с липидами.
Схема метода
1-й этап. 5 г гомогенизированного биологического материала четырехкратно извлекают ацетоном последовательно 10,0; 5,0; 5,0 и 5,0 мл с помощью аппарата для встряхивания, каждый раз в течение 5 мин. Затем пробы центрифугируют при 2,5 тыс. об./мин в течение 5 мин. Ацетоноводный экстракт смешивают с 4 г высушенного карбоната калия. Ацетоновый слой отделяют, добавляют 20 мл 0,5 М раствора хлороводородной кислоты и 10 мл н-гексана, встряхивают. Органическую фазу, содержащую гидрофобные примеси, отбрасывают.
2-й этап. Водную фазу экстрагируют эфиром, эфир испаряют и анализируют на вещества кислотного характера. Оставшийся водный раствор подщелачивают 25% раствором аммиака до рН=9-10, добавляют 3 г хлорида натрия (для высаливающего эффекта) и экстрагируют диэтиловым эфиром. Эфир испаряют и остаток исследуют на вещества основного характера. Оценка метода. Способ позволяет получать достаточно чистые экстракты. В процессе гомогенной экстракции анализируемые вещества практически не теряются. При реэкстракции потери веществ не превышают 5-6%.
6.4.2. Изолирование подкисленной водой
Этот метод является одним из наиболее эффективных и доступных. Соли многих лекарственных и наркотических веществ лучше растворяются в подкисленной воде, чем в подкисленном спирте.
Вода, подкисленная щавелевой кислотой, была предложена в 1856 г. С. Макадамом для выделения алкалоидов из биологических объектов. Для очистки извлечений был использован активированный уголь.
В 1865 г. Г. Драгендорф для подкисления объекта при изолировании алкалоидов предложил использовать серную кислоту. Однако способы дальнейшей обработки извлечений были слишком громоздки, и поэтому эти методы не получили широкого распространения.
В 1942 г. А.В.Степанов и М.Д.Швайкова разработали метод жстракТши алкалоидов подкисленной водой из растительных объектов (пищевых продуктов). Этот метод в 1947 г. был модифицирован и применен А. А.Васильевой для экстракции алкалоидов из свежих биологических объектов, после чего подкисленная вода стала широко использоваться в практике химико-токсикологического анализа при изолировании различных токсических веществ.
Эти методы получили дальнейшее развитие и совершенствование в работах многих ученых. В.Ф.Крамаренко и его учениками (В.И.Поповой, Б.В.Швыдким, З.С.Рокач др.) были изучены оптимальные условия экстракции алкалоидов из трупного материала и из водных растворов. Ими были установлены оптимальное значение pH для максимальной экстракции алкалоидов, условия разрушения комплекса алкалоидов с белками и изученььвлияние электролитов и их оптимальной концентрации на изолирование ядовитых веществ, способы осаждения белков и удаления их из полученных водных вытяжек. Процесс фильтрования был заменен центрифугированием. Определена пригодность метода при анализе как свежего, так и загнившего трупного материала.
Разработка и детальное изучение оптимальных условий экстракции позволили внести изменения и в ранее предложенные методы изолирования лекарственных и наркотических веществ.
Метод Степанова-Швайковой
Схема метода
1-й этап. Навеску растительного объекта массой 5 г заливают водой очищенной (1:12), подкисляют щавелевой кислотой до рН=2-2,5 и настаивают 2 ч. Водную вытяжку центрифугируют в течение 30 мин при 3000 об./мин. Прозрачную жидкость отделяют от осадка.
2-й этап. Центрифугат экстрагируют последовательно хлороформом при рН=2, затем при рH = 10 после подщелачивания 25% раствором аммиака.
Метод А.А.Васильевой
Схема метода
1-й этап. Навеску измельченного биологического объекта массой 100 г заливают водой очищенной (1:2), подкисляют щавелевой кислотой до рН=2-2,5 и настаивают 2 раза: 2 ч и 1 ч при периодическом взбалтывании. Водную вытяжку процеживают через двойной слой марли и центрифугируют. Надосадочную прозрачную жидкость отделяют.
2-й этап. Центрифугат экстрагируют последовательно хлороформом при значениях рН=2, затем при рН=10 после подщелачивания 25% раствором аммиака.
Оценка методов. Методы Степанова -Швайковой и Васильевой уменьшают время на выполнение анализа по сравнению с методом Стаса-Отто в 3-4 раза и позволяют изолировать меньшие количества ряда алкалоидов. Они не требуют затраты этилового спирта, но не пригодны при анализе сильно загнивших объектов.
Метод В.Ф.Крамаренко
Схема метода
1-й этап. Навеску измельченного объекта массой 100 г заливают 0,02 М раствором серной кислоты (рН=2,5) и настаивают 2 раза: 2 ч и 1 ч. Водные вытяжки центрифугируют, отделяют от осадка, насыщают сульфатом аммония и вновь центрифугируют. Центрифугат (рН=2,5) экстрагируют диэтиловым эфиром с целью очистки.
2-й этап. Водную фазу после подщелачивания до рН=8,5—9 раствором гидроксида натрия экстрагируют 4 раза хлороформом.
Оценка метода. В методе максимально учтены химические свойства алкалоидов и органических оснований. Поэтому условия изолирования и последующей экстракции оптимальны для определения алкалоидов.
Сравнение приведенных методов (см. табл. 4) позволяет провести выбор условий изолирования в зависимости от конкретных условий.
Таблица 4. Сравнительная оценка методов изолирования алкалоидов (по данным В.Ф.Крамаренко)
|
Алкалоид |
Процент изолирования по методу |
||
|
Стаса-Отто |
Васильевой |
Крамаренко |
|
|
Морфии |
18-21 |
21-24 |
47-51 |
|
Кодеин |
24-26 |
33-40 |
59-63 |
|
Кокаин |
17-19 |
25-32 |
53-56 |
|
Хинин |
14-18 |
23-41 |
63-69 |
|
Пахикарпин |
19-22 |
54-58 |
66-70 |
|
Стрихннн |
22-26 |
30-34 |
50-56 |
|
Сюполамин |
11-16 |
35-39 |
61-66 |
|
Бруцин |
29-32 |
27-33 |
51-55 |
|
Атропин |
22-27 |
31-37 |
59-64 |
|
Время анализа |
7-8 дней |
4-6 ч |
4-6 ч |
Метод В. И. Поповой
Схема метода
1-й этап. Измельченный объект массой 100 г заливают 80 мл 0,02 М раствора серной кислоты (рН=2-3) и настаивают 3 раза: 2 ч, 1 ч и 1 ч. Водную вытяжку процеживают через тройной слой марли и центрифугируют в течение 25-30 мин при скорости 3000 об./ мин. Надосадочную жидкость отделяют и очищают с помощью гель-хроматографии. С этой целью предложен гель сефадекс G-25. Барбитураты с колонки элюируют с помощью 0,02 М раствора серной кислоты.
2-й этап. Элюат подвергают концентрационному экстрагированию путем повторной экстракции хлороформом или диэтиловым эфиром.
Оценка метода. Метод обеспечивает хорошую очистку извлечений. Он применим при исследовании объектов, подвергшихся гнилостным изменениям, однако может использоваться для изолирования ограниченного числа веществ, в частности барбитуратов.
6.4.3. Изолирование подщелоченной водой
Впервые экстракцию подщелоченной водой предложил П.Валов в 1946 г. Для осаждения белков он применил вольфрамат натрия. Метод может быть пригоден при исследовании объекта на наличие некоторых веществ кислотного характера (салициловая и бензойная кислота, фенолы, нитрофенолы). Первоначально П.Валов этот метод использовал для изолирования барбитуратов из крови. В настоящее время известно несколько модификаций метода П. Валова. Мы приводим модификацию метода, предложенную М.Д.Швайковой и ее сотрудниками.
Метод П.Валова
Схема метода
1-й этап. Измельченный объект массой 100 г заливают 180 мл подщелоченной воды (рН=10) и настаивают в течение 30 мин. Щелочную вытяжку центрифугируют. Надосадочную жидкость (центрифугат) подкисляют серной кислотой до рН=2. В полученном растворе с помощью вольфрамата натрия при нагревании осаждают белки и смесь вновь центрифугируют.
2-й этап. Барбитураты из центрифугата экстрагируют диэтиловым эфиром при значении рН=2. Эфирный слой отделяют и проводят реэкстракцию водным раствором гидроксида натрия. Щелочной водный экстракт подкисляют серной кислотой до рН=2 и барбитураты извлекают равным объемом диэтилового эфира. Такая повторная экстракция позволяет получить чистые извлечения.
Оценка метода. Метод эффективен, позволяет выделить 50-90% барбитуратов и получить достаточно чистые извлечения. Недостатком метода является сорбция некоторых барбитуратов белками.
6.4.4. Изолирование наркотических и одурманивающих веществ из мочи твердофазной экстракцией
Этот метод является альтернативой жидкостной экстракции. Он основан на сорбции токсических веществ на синтетических смолах, модифицированных силикагелях, активированном углем и др. Авторами метода являются Т.Н.Бурыкина и Б.Н.Изотов. В качестве сорбента ими предложен полисорб-1 (сополимер стирола и дивинилбензола). Метод разработан для выделения, очистки и концентрирования наркотических и одурманивающих веществ из мочи. Ядовитые вещества сорбируются на полисорбе, а затем элюируются соответствующим элюентом.
Схема метода
Для изолирования веществ кислотного характера мочу (50 мл) подкисляют до рН=2 хлороводородной кислотой и смесь пропускают через колонку с сорбентом со скоростью 1-2 мл/мин. Для изолирования веществ основного характера к 50 мл мочи добавляют раствор аммиака до рН-8 -9 и также пропускают через колонку с сорбентом.
Вещества с кислотными и нейтральными свойствами, которые удерживаются гидрофобными группами сорбента, элюируют растворителями средней полярности. Оптимальными элюентами являются этилацетат или смесь ацетона и этилацетата. Вещества основного характера, удерживаемые катионообменными группами сорбента в протонированной форме, элюируются смесью хлороформа и изопропанола (9:1 или 4:1). Органические растворители (элюенты) удаляют при 40°С под током азота. Сухой остаток анализируют. Чувствительность метода находится в пределах 0,06-0,4 мкг вещества в 1 мл мочи.
Оценка метода. Метод сорбции (твердофазной экстракции) позволяет упростить методику подготовки объекта к анализу, обрабатывать до 5 и более проб мочи одновременно, повысить чувствительность обнаружения токсических веществ, получить более чистые извлечения, практически исключить присутствие в элюатах веществ, определяющих запах мочи.
В 50 мл мочи методом сорбции можно определить: морфина 20 мкг, кодеина 2-5 мкг, аминазина, дипразина, промедола, пиперазина 4-5 мкг, наркотина 10 мкг, амитриптилина 8-10 мкг, диазепама 5-10 мкг.
6.4.5. Экстракция органическими растворителями
Большую группу ядов составляют органические ядохимикаты, применяемые в сельском хозяйстве (пестициды). Наиболее часто в химико-токсикологической практике встречаются следующие пестициды:
— фосфорсодержащие пестициды — производные фосфорной, тиофосфорной, дитио- фосфорной и фосфоновой кислот: тиофос, трихлорметафос-3, карбофос, хлорофос;
— хлорорганические производные — гексахлорциклогексаы, гептахлор;
— производные карбачиновой кислоты — севин;
— органические соединения ртути — алкилртутные соли;
— синтетические пиретроиды.
В настоящее время не существует единого, универсального метода изолирования ядохимикатов из различных объектов, нет и общей схемы очистки полученных экстрактов. Даже среди определенной группы отдельные соединения могут изолироваться из объекта различными методами, хотя для большинства веществ этой группы общим свойством является растворимость в органических растворителях. Чаще всего рекомендуются индивидуальные методы изолирования пестицида для каждого объекта (воздух, пищевые продукты растительного происхождения, почва, кровь, моча, внутренние органы, масла и др.). Методы очистки пестицидов, выделенных из биологических объектов, также разнообразны. Среди них перегонка с водяным паром, реэкстракция, кристаллизация, хроматография в тонком слое сорбента, ионообменная хроматография и др.
Изолирование фосфорсодержащих пестицидов (тиофос, трихлорметафос-3, карбофос, хлорофос)
При изолировании первых трех пестицидов биологический объект смешивают с водой до кашицеобразной массы и настаивают с хлороформом или бензолом 3 раза по 4 и 2 ч. Экстракты объединяют, фильтруют, выпаривают досуха. Сухой остаток растворяют- в небольшом объеме органического растворителя и исследуют.
При изолировании хлорофоса схема метода включает 2 этапа:
1-й этап. Биологический объект заливают водой, подкисленной серной кислотой, до рН=2-2,5 и настаивают 3 раза по 2 и 1 ч. Водные вытяжки центрифугируют, надо- садочную жидкость отделяют.
2-й этап. Центрифугат экстрагируют хлороформом 30 мл 4 раза, растворитель упаривают и в остатке проводят обнаружение и количественное определение хлорофоса и его основного метаболита — дихлоруксусного альдегида.
Изолирование хлорорганических производных
Для изолирования гексахлорциклогексана навеску биологического объекта смешивают с водой до кашицеобразной массы, подкисляют раствором щавелевой кислоты до рН=2 и подвергают перегонке с водяным паром. Холодильник промывают диэтиловым эфиром, дистиллят экстрагируют диэтиловым эфиром. Объединенные эфирные вытяжки промывают водой, испаряют и остаток анализируют.
При отравлении гептахлором объектами исследования являются печень и почки. Навеску объекта смешивают с водой до кашицеобразной массы и настаивают с гексаном или эфиром 3 раза по 30 мин. Объединенные гексановые экстракты взбалтывают с насыщенным раствором сульфата натрия в 20% серной кислоте до получения бесцветной водной фазы (очистка) или обрабатывают безводным сульфатом натрия. Очищенные гексановые вытяжки анализируют.
Для выделения гептахлора из биологических жидкостей навеску крови или мочи экстрагируют диэтиловым эфиром или гексаном трижды. Экстракты объединяют и смешивают с безводным сульфатом натрия, органический растворитель испаряют и остаток исследуют на гептахлор.
Изолирование производных карбаминовой кислоты (севина)
Навеску объекта смешивают с водой до кашицеобразной массы и настаивают с бензолом 3 раза по 4 и 2 ч. Бензольные вытяжки объединяют и выпаривают досуха. Сухой остаток растворяют в спирте и анализируют на севин и его основной метаболит а-нафтол.
Изолирование синтетических пиретроидов
При отравлении синтетическими пиретроидами объектами исследования могут быть кровь, моча, почки, селезенка, желудок, тонкий кишечник. При направленном анализе изолирование включает два этапа:
1-й этап. Пиретроиды экстрагируют из объекта гексаном, петролейным эфиром или смесью гексана и ацетона 9:1 или 7:3 путем взбалтывания в течение 10 мин. Экстракт очищают от примесей ледяной смесью ацетон-вода (2:1). Водный раствор фильтруют.
2-й этап. Экстракцию пиретроидов из полученного фильтрата проводят с помощью н-гексана.
При ненаправленном анализе изолирование включает также два этапа:
1-й этап. Настаивают объект с водой, подкисленной хлороводородной кислотой, до рН=2-3, центрифугируют и отделяют надосадочную жидкость.
2-й этап. Центрифугат экстрагируют органическим растворителем (диэтиловым эфиром, хлороформом).
Изолирование органических соединений ртути
Объектами исследования при отравлениях могут быть печень, почки, моча и кровь. Изолирование соединений ртути включает следующие этапы:
1-й этап. Навеску объекта настаивают с 3 М раствором хлороводородной кислоты 2 раза по 30-60 мин, затем смесь центрифугируют.
2-й этап. Надосадочную жидкость экстрагируют хлороформом. Для изолирования соединений ртути из мочи вначале к объекту добавляют концентрированную хлороводородную кислоту и настаивают 30 мин. Затем проводят трехкратно экстракцию хлороформом.Для изолирования органических соединений ртути из крови к объекту добавляют раствор гидроксида натрия. Смесь нагревают на водяной бане 5-10 мин. Полученную однородную жидкость смешивают с концентрированной хлороводородной кислотой и настаивают 10-15 мин, затем центрифугируют. Из надосадочной жидкости соединения ртути экстрагируют хлороформом.
6.4.6. Экстракция водой в сочетании с диализом
С помощью этого метода изолируют:
— минеральные кислоты: серная, азотная, хлороводородная;
— щелочи: гидроксиды натрия, калия, аммиак и др.;
— ядовитые соли: нитраты, нитриты.
Такие исследования проводят в том случае, если предварительные испытания дают для этого основание или материалы дела указывают на возможность отравления указанными веществами.
Если в трупном материале минеральные кислоты или едкие щелочи после отравления образовали соответствующие соли, их обнаружение не проводится, так как некоторые соли в определенных количествах всегда содержатся в органах и тканях людей и животных.
Для изолирования объекты измельчают и добавляют очищенную воду до получения кашицеобразной массы, которую оставляют на 1-2 ч, а затем фильтруют. Для очистки полученных вытяжек применяют метод диализа.
Диализ — освобождение водных вытяжек, содержащих ядовитые вещества, от высокомолекулярных соединений при помощи полупроницаемой мембраны. Дтя проведения диализа между вытяжкой и водой очищенной помещают пористую перегородку — мембрану, поры которой проницаемы для ионов и молекул низкомолекулярных веществ, но не пропускают белки и макромолекулы. Распределение (концентрация) ионов ядовитых веществ по обе стороны мембраны определяется мембранным равновесием, которое было описано и теоретически объяснено Ф.Доннаном. Впервые метод диализа был применен Т.Грэмом в середине XIX в.
В качестве мембран для диализа используют перегородки из животного пузыря, пергаментной бумаги, пленки из нитро- или ацетилцеллюлозы (коллодий, целлофан). В настоящее время полупроницаемые мембраны можно изготовить с любой степенью проницаемости, т.е. с любым диаметром пор. Применяются также пористые металлические перегородки.
Недостатком метода является возможность прилипания белковых молекул (частиц) к стенкам пор и их коагуляции при соприкосновении с поверхностью мембраны. Поэтому при выборе мембраны учитываются свойства и характер примесей в вытяжках из объектов.
Ускорения процесса диффузии ионов при диализе можно достичь наложением электрического поля (электродиализ). Полученные диализаты подвергают основному исследованию на наличие кислот, щелочей, ядовитых солей.
Обнаружение и определение «металлических» ядов в биологических объектах возможно после разрушения органических веществ. Необходимость полной минерализации объекта связана с тем, что соли металлов в организме вступают во взаимодействие с белками и образуют иногда очень прочные соединения. В этом случае металлы почти не извлекаются из объектов обычными методами (настаиванием с различными извлекающими жидкостями) и не могут быть обнаружены с помощью известных реакций. Минерализация создает возможность получения соединений металлов, легко переводимых в ионное состояние. Известны два метода минерализации: «мокрая минерализация» и «сухое озоление».
6.5.1. Методы «мокрой минерализации»
Все методы мокрой минерализации сводятся к разрушению органических веществ с помощью окислителей в кислой среде. Окислителями при мокрой минерализации могут быть серная кислота и ее смеси с азотной, хлорной кислотами или концентрированным раствором пероксида водорода. Следует подчеркнуть, что все реактивы, применяемые для мокрого озоления, особо опасны. Они могут вызвать сильные ожоги, являться взрывоопасными (хлорная кислота, пероксид водорода), а в смесях с органическими веществами — пожароопасными.
Различают несколько модификаций метода «мокрого озоления»:
— минерализация серной и азотной кислотами;
— минерализация серной, азотной и хлорной кислотами;
— минерализация для обнаружения ртути в объекте (частный метод).
Минерализация серной и азотной кислотами
Минерализацию проводят в колбе Кьельдаля (рис. 4), в которую помещают 100 г измельченного объекта и 75 мл смеси равных объемов азотной, серной кислот и воды. Колбу Кьельдаля укрепляют на 2 см выше асбестовой сетки. Над колбой закрепляют делительную воронку с азотной кислотой, разбавленной водой очищенной в соотношении 1:1.
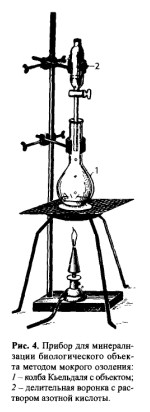
На первой стадии (деструкция) проводят медленное нагревание, не допуская обугливания. При этом разрушаются структурные элементы тканей и органические соединения, за исключением жиров. Окисление происходит с помощью азотной кислоты:
HNО3 + С (органические соединения) → NО2 + СО2 + Н2О.
Побочными реакциями на первой стадии могут быть реакции сульфирования и нитрования. Продукты этих реакций труднее минерализуются, поэтому для предотвращения их образования в начале минерализации рекомендуется добавлять воду.
По окончании первой стадии минерализации получается прозрачная жидкость желтоватого или буроватого цвета.
Для разрушения жиров (вторая стадия) колбу Кьельдаля опускают на асбестовую сетку, нагревание усиливают и начинают по каплям добавлять азотную кислоту из делительной воронки. На этой стадии окислительные свойства проявляет наряду с азотной кислотой и серная кислота, так как ее концентрация достигает 70% за счет испарения воды из колбы:
H2SО4 + С (органические соединения) → SО2 + СО2 + Н2О.
Если первая стадия заканчивается за 15-40 мин, то вторая стадия длится 3—4 ч. Окончанием этой стадии считается момент, когда не наблюдается обугливания минерали- зата при 30-минутном нагревании без добавления азотной кислоты. Минерализат при этом должен быть бесцветным, а при наличии меди или хрома может быть голубоватым или зеленоватым.
Для денитрации (удаление оксидов азота, азотной, азотистой кислот) к минерализату добавляют 10-15 мл воды очищенной, нагревают до 110- 130°С и по каплям в смесь вносят формалин. При этом происходят реакция восстановления азотной и азотистой кислот и окисление формальдегида по следующим уравнениям:
4HNO3 + 3CH2O → 4NO + 3CO2 + 5H2O
4HNO3 + 5CH2O → N2 + 2CO2 + 7H2O
4HNO3 + 2CH2O → N2 + 2NO+ 2CO2 + 4H2O
2NO + O2 → 2NO2
Реакция заканчивается за 1-2 мин. Возможные остатки формальдегида удаляются при нагревании жидкости в течение 5-10 мин. Проверку окончания денитрации проводят с помощью дифениламина в серной кислоте. Отсутствие синего окрашивания свидетельствует о полном удалении из минерализата соединений азота.
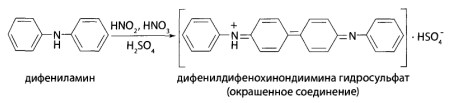
После завершения денитрации минерализат разбавляют водой очищенной до 180 мл. Если образуется осадок, жидкость нагревают до кипения для укрупнения осадка и охлаждают. Осадок отфильтровывают и промывают 1% раствором серной кислоты. Осадок анализируют на содержание ионов бария и свинца. Фильтрат разбавляют в мерной колбе водой очищенной до 200 мл и исследуют на катионы марганца, хрома, серебра, меди, висмута, цинка, таллия, сурьмы, мышьяка, кадмия и др.
Оценка метода. Метод позволяет быстро достичь полноты разрушения органических веществ. В результате получается малый объем минерализата, что увеличивает возможность обнаружения ядов с помощью известных реакций и методов. Недостатком метода является частичная или полная потеря ртути в процессе минерализации.
Минерализация серной, азотной и хлорной кислотами
Добавление хлорной кислоты в минерализирующую смесь ускоряет процесс окисления. В нашей стране метод применяется сравнительно редко и используется для минерализации особо стойких органических объектов. В разрушении органических веществ принимают участие все три кислоты: азотная, серная (по ранее приведенным схемам) и хлорная.
2НСlO4+ С (органические соединения) → Сl2О6 + СO2 + Н2O.
По технике исполнения метод аналогичен предыдущему. Минерализацию проводят в колбе Кьельдаля или в аппарате Бетге. Аппарат Бетге (см. рис. 5) используется при необходимости улавливания летучих продуктов окисления при минерализации. К измельченному объекту добавляют через воронку по 25 мл концентрированных серной и азотной кислот и 35 мл 37-42% хлорной кислоты.
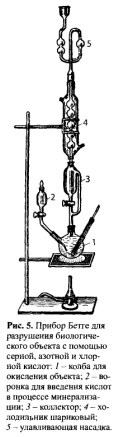
Нагревание усиливают постепенно, добавляя при обугливании по каплям концентрированную азотную кислоту. Когда минерализат станет прозрачным, его разбавляют водой очищенной (1:1) и проверяют полноту разрушения путем прибавления 25% раствора аммиака. При отсутствии оранжевого окрашивания (реакция на трудноокисляемые аминокислоты: триптофан, тирозин, фенилаланин) считают окисление объекта законченным.Оценка метода. Основным достоинством метода является более полное и быстрое окисление. Время минерализации сокращается в 2-3 раза по сравнению с методом разрушения серной и азотной кислотами. Потери ртути меньше, чем в предыдущем методе, так как разрушение происходит в замкнутой системе.
6.5.2. Метод минерализации для обнаружения ртути в объекте
При использовании общепринятых методов минерализации ртуть практически полностью теряется, так как ее соединения летучи. Поэтому делались многочисленные попытки усовершенствовать стадии минерализации объекта. В настоящее время в химико-токсикологическом анализе используется частичная (деструктивная) минерализация в модификации А. Н. Крыловой.
К объекту массой 20 г (печень или почки раздельно) добавляют 5 мл воды очищенной, 1 мл этилового спирта и 10 мл концентрированной азотной кислоты. Затем по каплям добавляют 10 мл концентрированной серной кислоты. Такой процесс позволяет контролировать скорость разложения азотной кислоты. Этому способствует и наличие этилового спирта, который способен связывать некоторое количество азотной кислоты непрочной эфирной связью. По окончании добавления серной кислоты колбу оставляют на 15 мин при комнатной температуре до прекращения выделения оксидов азота, а затем нагревают на водяной бане 10-20 мин. Если при нагревании реакция протекает слишком бурно с выделением бурых паров, то добавляют 30-50 мл горячей воды. Горячий деструктат фильтруют в колбу с 20 мл насыщенного раствора мочевины, который добавляют для проведения денитрации. В данном случае не требуется более сильных восстановителей, так как наличие этанола и условия проведения деструкции приводят в итоге к образованию азотной кислоты и оксидов азота низшей степени окисления.
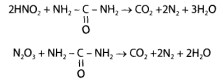
Избыток мочевины удаляют нагреванием в присутствии серной кислоты.
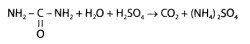
Деструктат разбавляют водой до определенного объема и далее проводят необходимые испытания.
Данный метод позволяет разрушить основные структурные элементы тканей (неразрушенными остаются жиры и трудноокисляемые аминокислоты). Связь ртути с белками разрушается.
Оценка метода. Метод позволяет достаточно быстро и полно разрушить связь ртути с белками. Потери ртути составляют не более 10% при ее содержании от 0,5 до 1,0 мг в 100 г объекта.
6.5.3. Методы «сухого озоления»
В химико-токсикологическом анализе из способов «сухого озоления» в ряде случаев применяют метод сплавления с карбонатом и нитратом натрия и метод простого сжигания. Их используют при небольших количествах объекта исследования, при отсутствии необходимости анализа на наличие соединений ртути и др.
Метод сплавления с карбонатом и нитратом натрия применяется и как дополнительный способ разрушения при специальных исследованиях на наличие в объекте мышьяка, сурьмы или для разделения свинца, бария, серебра. Особенно он удобен при анализе органических красителей, остатков мочи, волос, ногтей и т.п.
Метод простого сжигания используется при исследовании объекта на наличие солей меди, марганца, висмута, цинка, фторидов и кремнефторидов.
Метод простого сжигания при определении соединений меди, марганца, висмута и цинка
На исследование из объекта берут небольшие навески массой 1-10 г (пищевые продукты, биологический материал). Измельченные объекты высушивают на песчаной бане, затем обугливают, усиливая нагрев до 300-400°С. После охлаждения обугленный объект смачивают насыщенным раствором нитрата аммония и нагревают на слабом огне до образования белой золы. Остаток обрабатывают хлороводородной или азотной кислотами. Полученный раствор фильтруют, выпаривают досуха, остаток растворяют в воде и используют для исследования.
Метод простого сжигания при определении соединений фтора и кремнефторидов
К соединениям фтора, имеющим токсикологическое значение, относятся соли фтористоводородной кислоты (NaF, NaHF2, CaF2) и кремнефториды (Na2SiF6).
Для изолирования фторидов измельченные органы трупа, содержимое желудка, пищевые продукты и др. в количестве 25 г подщелачивают избытком гашеной извести или гидроксидом натрия, смачивают раствором нитрата аммония или концентрированной азотной кислоты, высушивают и прокаливают при температуре не выше 500°С до полного сжигания. Полученная зола подвергается анализу. При подозрении на возможность присутствия фторидов в реактивах и посуде необходимо провести в тех же условиях слепой опыт.
Метод сплавления с карбонатом и нитратом натрия
Навеску объекта массой 1-2 г растирают со смесью карбоната и нитрата натрия (2:1). В тигель помещают 5-6 г нитрата натрия, расплавляют его путем нагревания, а затем малыми порциями вносят подготовленную растертую массу объекта с карбонатом и нитратом натрия. Смесь нагревают на асбестовой тарелке, не допуская образования вспышек пламени в объекте. Сжигание считают законченным, когда исчезла черная окраска после добавления последней порции смеси. Остаток, представляющий собой белый сплав, обрабатывают горячей водой (выщелачивают). В водном растворе сплава определяют наличие ионов металлов.
При таком методе минерализации исключается обнаружение ртути, так как она способна улетучиваться не только в свободном состоянии, но и в виде некоторых солей.
6.6. Методы изолирования «летучих» ядов
Одним из методов изолирования ядовитых веществ является перегонка с водяным паром. С помощью этого метода из биологического материала изолируются следующие} группы веществ:
— алифатические спирты (алканолы): метиловый спирт, этиловый спирт, пропана бутанол, пентанол;
— диолы: этиленгликоль;
— алкилгалогениды: хлороформ, хлоралгидрат, четыреххлористый углерод, дихлорэтан;
— альдегиды: формальдегид;
— кетоны: ацетон;
— одноатомные фенолы и их производные: фенол, крезолы;
— карбоновые кислоты: уксусная кислота;
— синильная кислота;
— некоторые алкалоиды: никотин, анабазин, пахикарпин.
Перегонке с водяным паром могут подвергаться вещества независимо от их способности смешиваться с водой или других физических и химических свойств. Это видно из приведенных характеристик веществ:
— кристаллические: фенол, о-крезол, п-крезол, хлоралгидрат;
— газообразные: формальдегид, синильная кислота (выше температуры 26,5°С);
— жидкости, хорошо смешивающиеся с водой: синильная кислота, метиловый спирт этиловый спирт, этиленгликоль, ацетон;
— жидкости, плохо смешивающиеся с водой и имеющие плотность >1: дихлорэтан, четыреххлористый углерод, хлороформ; имеющие плотность — вещества, мало растворимые в воде: о-крезол, п-крезол, фенол, хлороформ, дихлор этан, четыреххлористый углерод;
— вещества, хорошо растворимые в воде: хлоралгидрат.
6.6.1. Метод перегонки с водяным паром
Перегонка с водяным паром в химико-токсикологическом анализе используется для выделения ядовитых веществ из различных объектов (рвотные массы, пищевые продукты, внутренние органы). Этот метод может использоваться для изолирования веществ как с низкой, так и с высокой температурой кипения.
В этом методе необходимо рассмотреть два случая: перегонка с водяным паром веществ, не смешивающихся с водой, и перегонка веществ, смешивающихся с водой.
В первом случае общее давление насыщенных паров равно сумме парциальных давлений каждого компонента:
Ро = Р1 + Р2
Парциальные давления воды и второго компонента не зависят друг от друга. Когда общее давление паров превышает атмосферное, начинается кипение смеси и интенсивное испарение обеих жидкостей. Температура кипения такой смеси ниже, чем температура кипения каждого компонента. Это является положительным моментом при перегонке веществ с высокой температурой кипения. В качестве примеров таких веществ можно назвать бензол, толуол, нитробензол, дихлорэтан, бутанол и др.
С помощью перегонки с водяным паром удается избежать слишком высоких температур, при которых некоторые вещества могут разлагаться. Например, тетраэтилсвинец разлагается при температуре выше 100°С. Его температура кипения равна 200°С, при которой он практически весь разлагается. При перегонке с водяным паром удается изолировать неразложившийся тетраэтилсвинец.
Перегонка с водяным паром используется для выделения из биологического материала жидкостей, которые с водой образуют растворы. Температура кипения таких смесей зависит от их состава. Чем больше жидкости с более низкой температурой кипения, тем ниже температура кипения смеси. Температура кипения смеси не может быть ниже той, при которой кипит более низкокипящая жидкость. Это наблюдается во всех случаях, когда две жидкости не образуют азеотропных смесей. Однако известно около 10000 систем, которые образуют азеотропные смеси. Это примерно половина всех изученных смесей. На кривых зависимости температуры кипения смеси от состава наблюдается максимум или минимум в точке, соответствующей азеотропной смеси. Среди двойных систем с азеотропными смесями примерно 93% приходится на системы с минимумом на кривой зависимости температуры от состава. Температура кипения азеотропной смеси ниже, чем любого компонента в отдельности.
В качестве примеров можно привести следующие смеси. Азеотропная смесь анилина (t° кипения 184,25°С) и воды (t° кипения 100°С) в соотношении 19,2% и 81,8% кипит при температуре 75,0°С. Азеотропная смесь толуола (t° кипения 110,7°С) и воды (t° кипения 100°С) в соотношении 81,4% и 19,6% кипит при температуре 84,1°С.
Однако встречаются азеотропные смеси, температура кипения которых выше, чем температура кипения каждого компонента. Примером такой азеотропной смеси является раствор хлороводородной кислоты. Раствор, содержащий 20,24% хлористого водорода (t° кипения — 85°С) и 79,76% воды (t° кипения 100°С), кипит при температуре 108,5°С.Иногда, для улучшения перегонки с водяным паром некоторых веществ, добавляется третий компонент, который в токсикологической химии принято называть «селективным переносчиком». Сущность такого приема заключается в том, что третий компонент повышает общую упругость паров в системе и этим самым позволяет снизить температуру кипения в системе. Третий компонент может образовывать с одним из компонентов азео- тропную смесь с пониженной температурой кипения. Так, при изолировании перегонкой с водяным паром этиленгликоля (t° кипения 197°С) в качестве «селективного переносчика» используют бензол. При этом температура перегонки снижается до 118°С. При перегонке уксусной кислоты (t° кипения 118°С) в присутствии гептана температура перегонки снижается до 80°С.
Таким образом, е токсикологической химии используются такие условия, при которых «летучие» яды изолируются в процессе перегонки при более низких значениях температуры, чем температура кипения каждого индивидуального вещества. Это особенно важно, так как при высокой температуре исследуемый объект может подгорать, что иногда приводит к образованию некоторых ядовитых соединений (например, синильной кислоты) и к ложно-положительным результатам анализа.
Перегонку с водяным паром проводят в специальном приборе, состоящем из нескольких частей (см. рис. 6).
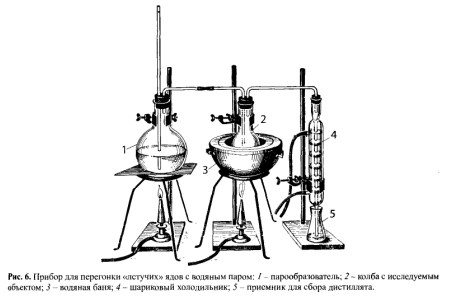
Парообразователь (1) представляет собой колбу, в которую вставлена пароотводная трубка и трубка-пеногаситель. Колба для объекта исследования (2) помещается в нагретую водяную баню (5) для предотвращения конденсации горячего пара. В шариковый холодильник (4) противотоком подается холодная вода. Колба (5) служит приемником для дистиллята. Обычно используют несколько приемников поочередно для сбора веществ разной степени летучести.
Методика проведения перегонки с водяным паром
Навеску объекта тщательно измельчают, смешивают с водой до густоты кашицы и помещают в колбу для перегонки. Соединяют колбу с приемником через холодильник. Конец холодильника опускают в жидкость, которая находится в приемнике при сборе первой порции дистиллята. С другой стороны присоединяют к колбе парообразователь. Парообразующую трубку погружают в кашицеобразную массу объекта в колбе почти до дна. Все соединения должны быть герметичны. Парообразователь с водой предварительно нагревают для выделения «сухого» пара.
Объект подкисляется до рН=2-2,5 раствором щавелевой кислоты. Подкисление необходимо, так как вещества кислотного характера перегоняются из кислой среды (например, фенол, синильная кислота). Применение для подкисления объекта минеральных кислот нежелательно, так как под влиянием минеральных кислот синильная кислота и ее соли разлагаются (реакция в присутствии серной кислоты и воды):
HCN = NH3 + HCOOH
Кроме того, при подкислении объекта минеральными кислотами фенол, поступивший в организм извне и вызвавший отравление, а также фенол, образовавшийся в кишечнике при разложении белковых молекул, будут одновременно перегоняться с водяным паром:
Если объект подкислен щавелевой кислотой, сернокислый эфир не гидролизуется и перегоняться будет только экзогенный фенол, который вызвал отравление.
При специальном исследовании объекта на уксусную кислоту подкисление проводят фосфорной или серной кислотами.
Сбор дистиллята
1-ю порцию дистиллята в количестве 3 мл собирают в 2 мл 5% раствора гидроксида натрия. Исследованиями И.В.Герасимова показано, что для сбора 1-й порции дистиллята лучше использовать в качестве поглощающего раствора смесь 2% растворов карбоната и гидрокарбоната натрия (1:1). При этом синильная кислота переходит в ее соль, и, таким образом, предотвращается ее потеря за счет высокой летучести:
HCN + NaОН = NaCN + Н2O.
Весь объем первой порции дистиллята исследуют только на синильную кислоту.
2-ю порцию дистиллята отгоняют в объеме 25-50 мл. Она содержит вещества средней степени летучести (спирты, ацетон, алкилгалогениды и др.).
3-ю порцию дистиллята также собирают в объеме 25-50 мл. В ней содержатся груднолетучие вещества (формальдегид, этиленгликоль и др.).
В процессе изолирования по внешнему виду дистиллята, его запаху можно предположить присутствие некоторых веществ, особенно, если их количество в дистилляте более 1 г (редко встречается в практике химико-токсикологическош анализа). Так, можно обнаружить маслянистые капли на поверхности дистиллята и ощутить характерный раздражающий запах сивушного масла (амиловые спирты), можно обнаружить тяжелые капли на дне дистиллята и ощутить характерный сладковатый запах при отравлении хлороформом, дихлорэтаном, четыреххлористым углеродом. Присутствие в дистилляте фенола придает ему характерный запах карболовой кислоты, молочновидное помутнение, и могут появиться розоватые капли на дне приемника за счет продуктов окисления фенола.
Поскольку некоторые «летучие» яды малорастворимы в воде, их после дистилляции извлекают органическим растворителем — диэтиловым эфиром или хлороформом. Так, перед обнаружением амиловых спиртов дистиллят экстрагируют диэтиловым эфиром и после его испарения с остатком проводят реакции на амиловые спирты. Для извлечения фенола из дистиллята вначале добавляют гидрокарбонат натрия, который связывает летучие кислоты, а затем экстрагируют диэтиловым эфиром непрореагировавший с гидрокарбонатом фенол.
Выделение некоторых алкалоидов перегонкой с водяным паром
Метод используется для изолирования в виде оснований жидких алкалоидов — анабазина, никотина, пахикарпина. Измельченный и смешанный до кашицеобразной массы с водой объект подщелачивают 20% раствором карбоната натрия и подвергают перегонке с водяным паром. Дистиллят собирают в 5% раствор хлороводородной кислоты, который по окончании перегонки подщелачивают раствором аммиака до рН=9-10 и алкалоиды- основания экстрагируют органическим растворителем (хлороформом).
6.6.2. Методы «микроперегонки» и микродиффузии
Метод «микроперегонки» состоит в образовании парогазовой фазы без дальнейшей ее конденсации. В герметически закрытый флакон помещают 1-5 г биологического объекта, добавляют сильный электролит и подогревают. Через некоторое время равновесная парогазовая фаза отбирается микрошприцем и используется для анализа с помощью газожидкостной хроматографии. Метод не позволяет получить полноту отгонки вещества. Он предложен для определения легколетучих спиртов, которые обнаруживаются этим методом за счет их низкой температуры испарения.
Метод микродиффузии используется при целенаправленном анализе. С помощью этого метода можно обнаружить «летучие» яды, содержащиеся в небольших количествах в исследуемом объекте. Для ускорения перехода отдельных веществ из объекта в пространство прибора используют электролиты. Например, прибавление насыщенного раствора карбоната калия к крови, моче, гомогенату тканей, содержащих этиловый спирт, ускоряет его переход в пространство прибора, для других соединений добавляют кислоты, щелочи и др.
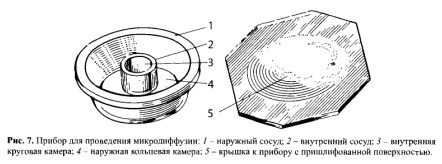
Прибор для микродиффузии (рис. 7) состоит из круглого тонкостенного сосуда (1), внутри которого расположен второй сосуд меньшего диаметра (2). Большой сосуд закрывается герметично пришлифованной крышкой (5). Исследуемый объект в количестве до 1 г (1 мл) вносят в круглый тонкостенный сосуд большего диаметра (/), а поглощающую жидкость — в сосуд меньшего размера (2). К объекту добавляют электролит, сосуд плотно закрывают крышкой и оставляют на определенное время, необходимое для диффузии вещества. Летучие вещества из объекта переходят в пространство прибора, а затем в поглотительный раствор сосуда меньшего диаметра. В качестве поглощающей жидкости используют соответствующий растворитель или раствор реактивов, способных реагировать с обнаруживаемым веществом. Время диффузии при анализе биологических жидкостей (крови, мочи) составляет 3-4 ч, гомогената тканей — 5-12 ч.
Для формальдегида, ацетона, альдегида уксусной кислоты в качестве поглотительных растворов используют 0,15 М раствор гидросульфита или сульфита натрия; для метилового спирта — 10% раствор серной кислоты; для этилового спирта — раствор дихромата калия в серной кислоте; для сульфидов, уксусной кислоты, фенола и цианидов — 0,1 М раствор гидроксида натрия; для оксида углерода (II) — 0,1% раствор хлорида палладия в 0,1 М растворе хлороводородной кислоты.
По окончании времени, необходимого для проведения анализа, с полученной поглощающей жидкостью из сосуда с меньшим диаметром проводят соответствующие реакции. Формальдегид обнаруживают по образованию розового или фиолетового окрашивания с хромотроповой кислотой; ацетон — по реакции с салициловым альдегидом (красное окрашивание); сульфиды — по реакции с солями висмута (осадок коричневого цвета); цианиды — по реакции образования полиметинового красителя; оксид углерода (II) — по образованию серебристой пленки металлического палладия и т.п.
Оценка метода. Метод дистилляции отличается простотой и экономичностью. При дистилляции получаются чистые извлечения-дистилляты. Метод микроперегонки используется только для обнаружения веществ. Метод микродиффузии является предварительным, ему можно придать судебно-химическое значение при отрицательном результате.