Фармацевтические субстанции (ОФС.1.1.0006.15)
Государственная фармакопея 13 издание (ГФ XIII)
Взамен ГФ XII, ч. 1, ОФС42-0074-07
Фармацевтические субстанции — лекарственные средства в виде одного или нескольких обладающих фармакологической активностью действующих веществ вне зависимости от природы происхождения, предназначенные для производства, изготовления лекарственных препаратов и определяющие их эффективность.
Требования данной статьи распространяются преимущественно на фармацевтические субстанции химического и минерального происхождения. Для субстанций, представляющих собой стандартизованную смесь биологически активных веществ растительного или животного происхождения возможны отклонения от данных требований или дополнительные требования, указанные в фармакопейных статьях.
Требования данной статьи распространяются также на вспомогательные вещества, используемые при производстве/изготовлении лекарственных препаратов.
В качестве названия фармакопейной статьи на фармацевтическую субстанцию используется общепринятое название. Многие субстанции представляют собой соли органических кислот и органических оснований (например, Кеторолака трометамол, или Амлодипипа бесилат, или Доксазозина мезилат), органических кислот и неорганических оснований (например. Диклофенак натрия), неорганических кислот и органических оснований (например, Кетамина гидрохлорид). Названия фармакопейных
Во вводной части фармакопейной статьи на субстанцию приводят химическое название по номенклатуре IUPAC, структурную формулу, брутто -формулу и относительную молекулярную массу
Вводимые показатели контроля качества и пределы нормирования должны соответствовать назначению субстанции (например, для производства/изготовления стерильных лекарственных препаратов, или стерильных неиньекционных лекарственных препаратов, или нестерильных лекарственных препаратов, или нестерильных лекарственных препаратов для местного и наружного применения и т.д.).
Испытания по показателям контроля качества фармацевтической субстанции проводят согласно соответствующим общим фармакопейным статьям (ОФС).
Описание.
Указывают характеристики физического состояния и цвет субстанции. Не следует включать описание вкуса. В необходимых случаях приводят информацию о запахе, гигроскопичности и полиморфизме.
Для твердых субстанций необходимо указание формы вещества: «кристаллический», «мелкокристаллический» или «аморфный порошок». Характеристика кристалличности субстанции является одним из важных параметров, от которого зависит качество твердых дозированных лекарственных препаратов.
В некоторых случаях может быть указан численный диапазон размера частиц, а также введено исследование формы кристаллов. Такие испытания выносят в отдельные разделы.
Оценка полиморфизма субстанции обязательна в тех случаях, когда полиморфная модификация определяет фармакологическую активность лекарственного препарата и его фармако-технологические свойства.
Растворимость.
Для определения растворимости следует использовать растворители, охватывающие широкую шкалу полярности, например: вода, спирт 96 %, гексан и др. Не рекомендуется использование легкокипящих и легковоспламеняющихся (например, диэтиловый эфир) или очень токсичных (например, бензол) растворителей.
Подлинность.
Для установления подлинности субстанции рекомендуются физико-химические и химические методы — инфракрасная спектрометрия, абсорбционная спектрофотометрия, ЯМР-спектроскопия, тонкослойная, газовая и высокоэффективная жидкостная хроматография (ТСХ, ГХ и ВЭЖХ) и качественные (в первую очередь специфические) химические реакции. Метод ИК-спектрометрии является приоритетным при идентификации субстанций.
Температура плавления.
Испытание обычно применяют для характеристики твердых веществ.
Температура затвердевания. Температура кипения (температурные пределы перегонки), Плотность, Вязкость, Показатель преломления.
Данные испытания вводят для характеристики жидких субстанций.
Удельное вращение.
Вводят для характеристики оптически активных веществ.
Удельный показатель поглощения.
Данный показатель может являться дополнительной характеристикой подлинности и чистоты субстанции.
Прозрачность раствора, Цветность раствора.
Данные испытания обязательно вводят для субстанций, используемых для приготовления парентеральных, глазных, назальных и ушных лекарственных средств. Испытание обычно проводят в водных растворах субстанции, но возможно использование органических и смешанных растворителей. Концентрация испытуемых растворов должна быть приближена к концентрации производимого/изготавливаемого из этой субстанции лекарственного препарата.
Определение цветности раствора особенно важно для оценки качества белых, почти белых или белых с оттенком субстанций.
Если субстанция окрашена, показатель «Цветность раствора» в нормативную документацию включать не следует. Это испытание, если необходимо, можно заменить регламентацией оптической плотности при определенных длинах волн.
pH и Кислотность или Щелочность.
Для проведения данного испытания могут использоваться два подхода: измерение pH или кислотно-основное индикаторное титрование (кислотность или щелочность). Испытание обычно проводят в водных растворах субстанции, но в отдельных случаях возможно использование и смешанных растворителей. Допустимый интервал pH обычно должен быть не более 2.
Концентрация испытуемого раствора при определении pH должна быть приближена к концентрации изготавливаемого из субстанции лекарственного препарата.
Родственные примеси
Данное испытание контролирует продукты деструкции фармацевтической субстанции и технологические примеси, обусловленные технологией производства. Примеси могут быть идентифицированные (соединения с установленным химическим строением) и неидентифицированные (соединения, строение которых не установлено). Пределы содержания родственных примесей в фармацевтических субстанциях приводят с учетом параметров их безопасности. Пределы контроля, идентификации и квалификации родственных примесей для фармацевтических субстанций (в зависимости от максимальной суточной дозы лекарственного препарата) приведены в табл. 1 и 2.
Таблица 1. Пределы контроля, идентификации и квалификации родственных примесей в фармацевтических субстанциях
* предел, выше которою примесь должна контролироваться
** предел, выше которого примесь должна быть идентифицирована
***предел, выше которого должна быть установлена биологическая безопасность примеси
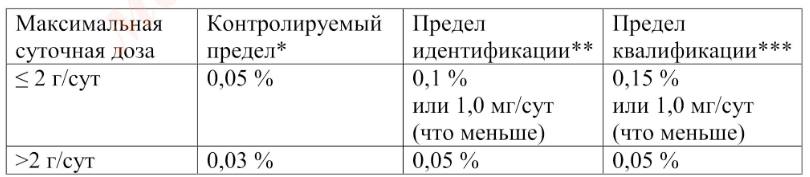
Приведенные пределы учитываются при нормировании родственных примесей в фармацевтических субстанциях.
Таблица 2. Пределы контроля, идентификации и квалификации родственных примесей в пептидах, полученных синтетическим путём
| Контролируемый предел | Предел
идентификации |
Предел
квалификации |
| >0,1 % | > 0,5 % | > 1,0% |
Для контроля родственных соединений обычно используют хроматографические и, реже, спектроскопические методы. Обязательно вводится идентификация и количественное определение токсичных примесей с использованием стандартных образцов.
Неорганические анионы (хлориды, сульфаты и др.).
Выбор контролируемых анионов определяется технологией получения субстанции. При этом контролируемые анионы могут быть нетоксичными (например, хлориды, сульфаты и т.д.).
Контроль анионов не вводят, если они входят в состав субстанции (например, субстанция является гидрохлоридом или сульфатом).
Неорганические катионы (железо, медь и др.).
Это испытание вводят, если контроль содержания отдельных катионов является существенным для качества субстанции; их содержание должно быть обосновано.
Контроль катионов не вводят, если они входят в состав субстанции (например, вещество является натриевой солыо).
Потеря в массе при высушивании или Вода.
Испытание вводят для контроля содержания летучих веществ и/или влаги в субстанции. Введение одного из этих испытаний, как правило, обязательно. Отсутствие их должно быть обосновано. Если нет других указаний в фармакопейной статье и субстанция не является кристаллогидратом (кристаллосольватом), потеря в массе при высушивании или содержание воды, как правило, не должно превышать 0,5 %. Результаты определения по этим показателям учитывают при оценке результатов количественного определения.
Если субстанция является кристаллогидратом (кристаллосольватом), регламентируют верхний и нижний пределы.
Сульфатная зола.
Как правило, сульфатная зола не должна превышать 0,1 %. Отсутствие этого испытания в фармакопейной статье или повышенное содержание сульфатной золы требует соответствующего обоснования.
Тяжелые металлы.
Устанавливаемые пределы содержания тяжелых металлов в фармацевтических субстанциях определяются максимальной суточной дозой препарата, произведенного из данной субстанции, и длительностью его возможною применения (согласно Инструкции по медицинскому применению) (табл. 3).
Мышьяк.
Данное испытание вводят в том случае, когда или исходное сырье может содержать мышьяк, например, для сырья природного происхождения, или возможно загрязнение им в процессе получения субстанции. Содержание мышьяка, как правило, не должно превышать 0,0001 %.
Таблица 3. Критерии для нормирования допустимого содержания тяжелых металлов
| Суточная доза, г/день | Длительность лечения, дни | Введение показателя «Тяжелые металлы» и устанавливаемый предел, ppm |
| >0.5 | <30 | Вводится показатель «Тяжелые металлы», предел — 20 |
| >0,5 | >30 | Вводится показатель «Тяжелые металлы», предел — 10 |
| < 0.5 | >30 | Вводится показатель «Тяжелые металлы». Если субстанция предназначена для производства парентеральных препаратов, то предел — 10, в других случаях — 20. |
| < 0,5 | < 30 | Показатель «Тяжелые металлы» не вводится |
Остаточные органические растворители.
В фармацевтических субстанциях определяют остаточные количества органических растворителей 1 касса токсичности, если обосновано их применение, независимо от стадии, на которой они используются.
Для растворителей 2 класса токсичности, используемых не на последней стадии производства фармацевтических субстанций, в нормативной документации должен быть предусмотрен контроль их остаточного содержания, либо должно быть приведено обоснование отсутствия контроля их остаточного содержания; если растворители 2 класса токсичности используются на последней стадии, каждый из них должен быть определен количественно.
В фармацевтических субстанциях контроль растворителей 3 класса токсичности необходим, если они используются на последней стадии производства. Для определения остаточных количеств растворителей 3 класса токсичности, если их содержание не превышает 0,5 %, допускается применение неспецифического метода «Потеря в массе при высушивании»; при наличии растворителей 3 класса, если их содержание превышает 0,5 %, каждый из них должен быть определен количественно.
Испытания на бактериальные эндотоксины, пирогенность, аномальную токсичность, гистамин и/или депрессорные вещества не распространяются на вспомогательные вещества.
Бактериальные эндотоксины или Пирогенность.
Данные испытания проводят для субстанций, предназначенных для приготовления лекарственных форм для парентерального применения. Субстанции должны выдерживать тест на бактериальные эндотоксины или пирогенность без проведения предварительной стерилизации.
Если доказано, что субстанция не обладает пирогенными свойствами и в процессе производства не может быть загрязнена пирогенными примесями ые бактериальной природы, то следует проводить испытание на «Бактериальные эндотоксины».
Включение показателей «Пирогенность» и «Бактериальные эндотоксины» на альтернативной основе нецелесообразно ввиду различной чувствительности методов.
Аномальная токсичность
Испытанию на аномальную токсичность подлежат субстанции, получаемые из крови, органов, тканей человека или животного, растительного сырья, микроорганизмов и продуктов их жизнедеятельности, предназначенные для производства лекарственных препаратов для парентерального применения.
Гистамин и/или Депрессорные вещества
Испытанию на гистамин и депрессорные вещества подлежат субстанции, которые используются для приготовления лекарственных препаратов, предназначенных только для внутрисосудистого введения, и если в их составе могут быть изначально или приобретаются в процессе производства примеси, обладающие депрессорным действием (субстанции микробиологического или животного происхождения).
Микробиологическая чистота.
Уровень микробиологической чистоты субстанции должен обеспечивать уровень чистоты лекарственного препарата при его производстве/изготовлении из этой субстанции.
Стерильность.
Данное испытание вводят для субстанций, используемых в производстве готовых стерильных лекарственных средств, которые не подвергаются процедуре стерилизации.
Количественное определение.
Для количественною определения действующего вещества субстанции используют физико-химические и химические методы анализа.
Физико-химические и химические методы анализа для определения содержания действующего вещества в субстанции следует применять в сочетании с современными физико-химическими методами анализа, используемыми для идентификации субстанции и контроля примесей (ИК-спектрофотометрия, ВЭЖХ, ГХ и др.).
В случае солей обычно достаточно анализа только одного из ионов — предпочтительно фармакологически активного.
Содержание действующего вещества дается в пересчете на сухое вещество, если определяется потеря в массе при высушивании, в пересчете на безводное вещество, если определяется вода, в пересчете на безводное и свободное от остаточных органических растворителей вещество, если определяется вода» и остаточные органические растворители.
Упаковка и хранение.
Упаковка и условия хранения должны обеспечивать качество субстанции в течение установленного срока годности.
Маркировка.
Должна включать торговое и международное непатентованное наименование, информацию о назначении субстанции, наименование производителя, количество, условия хранения, меры предосторожности (при необходимости), дату изготовления, номер серии, срок годности и условия хранения.
Срок годности.
Срок годности субстанций определяется временем, в течение которого она соответствует требованиям нормативной документации. Срок годности субстанции может быть установлен хранением при обычных условиях или методом «ускоренного старения» при повышенной температуре.
Стандартные образцы.
Современные методы анализа предусматривают использование стандартных образцов. В качестве стандартных образцов при анализе фармацевтических субстанций следует использовать фармакопейные стандартные образцы, аттестованные уполномоченным фармакопейным органом. При их отсутствии для идентификации и оценки содержания действующего вещества должны использоваться первичные стандартные образцы.