ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Аналитическая химия. Васюкова А., Веденяпина М.– 2019
- 4.1. Основные понятия
- 4.2. Методы нахождения неизвестной концентрации
- 4.3. Метрологические характеристики физико-химических методов анализа
- 4.4. Анализ веществ по их физическим константам
4.1. Основные понятия
Качественная реакция или свойство вещества, которые можно использовать для получения информации о химическом составе вещества, называют аналитическим сигналом.
Все методы аналитической химии основаны на получении и измерении аналитического сигнала (АС). Для его получения используют химические реакции различных типов (кислотно-основные, окислительно-восстановительные, комплексообразования), различные процессы (осаждение, растворение, экстракция) и свойства анализируемых веществ или продуктов реакции (химические, физические, биологические).
В аналитической химии используются все возможности для получения информации о составе вещества, поэтому для современной аналитической химии характерно большое разнообразие методов анализа. Источником информации для химика-аналитика является проба. Измеренный АС пробы складывается из значимых, мешающих и шумовых сигналов.
Значимые — это полезные сигналы. При необходимости их надо усилить, используя концентрирование.
Мешающие — сигналы от растворителя, реагентов, примесей. Они накладываются на значимые сигналы, сливаются с ними, поэтому обнаруживаются одновременно с ними. Их необходимо устранить.
Шумовые — сигналы, которые не имеют отношения к анализируемому веществу. Они связаны с работой отдельных узлов приборов и электросети. Их надо снизить.
Полезный АС = Измеренный АС пробы – АС фона.
Для получения АС, наиболее близкого к истинному, используют различные приемы. Главным приемом является предварительное разделение. Для этого определяемый компонент выделяют в чистом виде. Для учета мешающих сигналов используется холостая проба (холостой опыт), которая содержит все компоненты, кроме определяемого. Она должна быть проведена через все стадии анализа. Сигнал холостой пробы вычитается из общего сигнала.
Количественный анализ вещества любым инструментальным методом основан на зависимости величины АС от концентрации определяемого компонента. Эта зависимость в общем виде может быть представлена как^

Методы нахождения неизвестной концентрации
Метод градуировочного графика — графический прием нахождения неизвестной концентрации (Сх) по величине аналитического сигнала пробы (Iх). Для проведения анализа готовят серию стандартных растворов, измеряют величины АС этих растворов и строят градуировочный график I = f(C) (рис. 4.1).
Требования к градуировочному графику:
- 1) график должен быть линеен, так как нелинейность снижает точность проведения анализа;
- 2) желательно, чтобы график выходил из начала координат;
- 3) график следует периодически проверять, а при замене каких-либо реагентов, растворов, приборов, условий проведения анализа построить заново;
- 4) в случае большого разброса точек надо применять метод наименьших квадратов, а не строить график «на глаз», особенно при работе с малыми концентрациями.

Рис. 4.1. Градуировочный график
Метод стандартов — расчетный прием нахождения неизвестной концентрации. Он имеет две разновидности. В методе одного стандарта для проведения анализа готовят один стандартный раствор с концентрацией определяемого вещества Сст, затем измеряют величины АС этого раствора (Iст) и пробы (Ix) в одинаковых условиях. Исходя из того, что Iст = k·Сст, находим k = Iст / Сст и рассчитываем неизвестную концентрацию:
Сх = Iх / k.
В методе двух стандартов для проведения анализа готовят стандартные растворы с концентрацией С1 и С2, измеряют величины АС этих растворов I1 и I2 и две пробы Ix в одинаковых условиях. Выбирают два стандартных раствора (ограничивающие растворы) так, чтобы:
![]()
Расчет неизвестной концентрации проводят по формуле:

Оба варианта метода можно применять только в том случае, когда зависимость I = f(С) является линейной.
Сущность метода добавок заключается в том, что сначала измеряют АС пробы (Iх), а затем АС той же пробы с добавкой стандартного раствора определяемого вещества (I х+ст). Метод имеет две разновидности.
Метод однократной добавки является расчетным. Неизвестную концентрацию С х рассчитывают по формуле:

Метод серии добавок является графическим. Для проведения анализа измеряют величины АС пробы и нескольких растворов той же пробы с добавками стандартного раствора. Строят график в координатах «Аналитический сигнал — концентрация добавки» (рис. 4.2) и по нему находят С х как величину отрезка, отсекаемого прямой на оси абсцисс. Метод добавок можно применять только в том случае, когда зависимость I = f (C) является линейной. Чаще всего метод добавок используют при анализе проб сложного состава, так как прирост АС при добавке стандартного раствора связан только с определяемым компонентом, а сигналы от мешающих компонентов пробы остаются постоянными.

Рис. 4.2. Построение графика методом серии добавок
В химических методах анализа аналитический сигнал появляется в ходе химических реакций.
Физико-химические методы анализа (ФХМА) — группа методов, основанных на регистрации изменения физических свойств, возникших в результате проведенных химических реакций. В физических методах анализа регистрируется аналитический сигнал без наличия химических реакций.
Физико-химические (как и химические) методы анализа основаны на проведении той или иной химической реакции. В физических методах химические реакции отсутствуют или имеют второстепенное значение. Поэтому их включают в группу физико-химических методов, так как достаточно строгого однозначного различия между физическими и физико-химическими методами нет и выделение их в отдельную группу не имеет принципиального значения.
При выполнении анализа физико-химическими методами точку эквивалентности (конец реакции) определяют не визуально, а при помощи приборов, которые фиксируют изменение физических свойств исследуемого вещества в точке эквивалентности. Для этой цели обычно применяют приборы с относительно сложными оптическими или электрическими схемами, поэтому эти методы получили еще название методов инструментального анализа.
Химические методы анализа, в отличие от инструментальных, являются безэталонными и позволяют непосредственно определять содержание вещества в пробе. В инструментальных методах необходимо провести предварительную калибровку шкалы приборов с помощью эталонов. Эталоны — это образцы, состав которых точно известен. При проведении анализа с использованием инструментального титрования измеряют какое-либо свойство раствора в процессе титрования. Кривые титрования (рис. 4.3) получаются разными в зависимости от измеряемой величины. Линейные кривые титрования получают, если АС линейно меняется при изменении концентрации вещества в растворе. К таким сигналам относятся, например, светопоглощение, сила тока, электрическая проводимость и др. Объем в точке эквивалентности (т.э.) находят по излому кривой.

Рис. 4.3. Кривая титрования
Логарифмические кривые титрования получают, если АС связан с логарифмом концентрации вещества в растворе. К таким сигналам относятся, например, потенциал, рН и др. Точку эквивалентности находят по перегибу кривой. В любом случае после построения кривой титрования и определения с ее помощью объема раствора титранта, который потребовался для достижения точки эквивалентности, расчет результатов анализа проводят по закону эквивалентов.
4.3. Метрологические характеристики физико-химических методов анализа
Важнейшими метрологическими характеристиками физических и физико-химических методов анализа являются чувствительность; предел определения; точность; правильность; воспроизводимость и селективность.
Чувствительность метода характеризует изменение АС при изменении концентрации вещества чем больше меняется величина сигнала, тем чувствительнее метод. Количественной мерой чувствительности служит коэффициент чувствительности S:
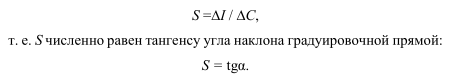
Чувствительность инструментальных методов анализа гораздо выше чувствительности классических методов из-за применения регистрирующих приборов.
Предел определения — минимальная концентрация, которую можно определить данным методом:

В инструментальных методах анализа предел определения в зависимости от метода составляет 10–5 –10–10 %, что соответствует содержанию вещества в пробе на уровне 10-6 ––10-15г. Предел определения в химических методах анализа обычно превышает 10-3 %.
Точность — это собирательная характеристика метода, включающая его правильность и воспроизводимость. Она отражает относительную погрешность (ошибку) определения (%). Обычно более чувствительные методы имеют меньшую точность, поскольку их используют при очень низких концентрациях. В связи с этим точность инструментальных методов анализа (более чувствительных) составляет в среднем 2–5 %, а химических методов (менее чувствительных) — 0,1–0,5 %.
Наиболее высокой точностью (до 0,001 %) обладает кулонометрия, основанная на измерении количества электричества, которое затрачивается на электрохимическое окисление или восстановление определяемых ионов или элементов. Однако такое сравнение погрешностей не вполне корректно, так как относится к разным концентрационным областям. При небольшом содержании определяемого компонента (около 10 –3 % и менее) классические химические методы анализа вообще непригодны.
Правильность характеризует близость полученного и истинного значений АС. Количественной мерой правильности служит систематическая погрешность. В инструментальных методах анализа, в отличие от химических, использование приборов является дополнительным источником систематических погрешностей.
Воспроизводимость отражает случайные ошибки измерения и показывает степень разброса параллельных (повторных) измерений. В инструментальных методах анализа, в отличие от химических, на воспроизводимость результатов дополнительно влияет стабильность работы приборов.
Селективность характеризует возможность определения нужного компонента без помех со стороны других компонентов пробы. Различные инструментальные методы анализа имеют разную селективность. Фотометрия пламени является высокоселективным методом, рефрактометрия — неселективным, поэтому ее используют только при анализе индивидуальных веществ или простых смесей, состоящих из 2–3 компонентов.
4.4. Анализ веществ по их физическим константам
Важными физическими константами индивидуальных веществ, которые используются для идентификации веществ и их смесей, являются:
- 1) температура плавления — отвердевания;
- 2) температура кипения — конденсации;
- 3) плотность;
- 4) показатель преломления.
Температура плавления вещества — это температура фазового перехода твердая фаза ↔ расплав. Ее определяют как при плавлении вещества, так и при кристаллизации расплава. Наиболее точным методом определения температуры плавления является метод термического анализа, основанный на измерении зависимости температуры нагреваемого вещества от времени при строго постоянной скорости нагревания или охлаждения. При этом не требуется непосредственное наблюдение за веществом. Метод в равной степени можно применять для определения как наиболее низких, так и очень высоких температур плавления.
Метод заключается в построении кривых нагревания или охлаждения в координатах «температура вещества — время» по данным визуального отсчета температуры или непосредственной записи кривых на саморегистрирующих приборах (пирометрах, дериватографах и др.). Рекомендуется сначала понаблюдать за поведением исследуемого вещества на кончике шпателя в пламени газовой горелки не разлагается ли оно до плавления, не взрывается ли, при какой примерно температуре плавится. В большинстве случаев скорость нагревания выбирают в интервале 5–10 °Смин, руководствуясь величиной навески вещества.
Капиллярный метод пригоден только для тех веществ, температуры плавления которых меньше 300 °С. Для наблюдения за температурой плавления вещества применяют сухие, чистые, тонкостенные капилляры с внутренним диаметром 0,5–1,0 мм. Заполненный веществом капилляр плотно прикрепляют к термометру резиновым кольцом так, чтобы столбик порошка в капилляре был около центра ртутного резервуара термометра. Затем термометр с капилляром погружают в колбу с нагреваемой жидкостью. Температуры плавления гигроскопичных и возгоняющихся веществ, а также веществ, чувствительных к действию воздуха, определяют в капиллярах, запаянных с двух концов. При этом такой капилляр должен быть весь погружен в нагреваемую жидкость.
Значения температуры отвердевания и плавления совпадают только для чистых веществ (табл. 4.1). Температуру плавления при нормальном атмосферном давлении (760 мм рт. ст.) называют точкой плавления. Смеси веществ не имеют температуры плавления, а совершают переход в диапазоне температур. Аморфные твердые тела переходят в жидкое состояние постепенно, размягчаясь с повышением температуры. Наличие температуры плавления у вещества — важный признак его кристаллического строения.
Таблица 4.1. Температура плавления некоторых веществ, °С

Температура кипения вещества при атмосферном давлении приводится обычно как одна из основных характеристик химически чистых веществ (табл. 4.2). Бурное образование пузырьков во всем объеме и называется кипением, а температура, при которой происходит процесс — точкой кипения. Определить температуру кипения (Т кип ) можно на перегонной установке. Она зависит от атмосферного давления. Например, на высоте 4000 м над уровнем моря вода кипит при 85 °С.
Таблица 4.2. Температура кипения некоторых веществ, °С (при 1 атм)

Плотность — физическая величина, определяемая для однородного вещества его массой в единице объема (при температуре 20 °С):
![]()
Измерение плотности вещества проводят с помощью ареометров (рис. 4.4, 4.5), пикнометров (рис. 4.6).


Плотность вещества различна в разных агрегатных состояниях (в твердом, жидком и газообразном). Плотность твердых тел чаще всего выше плотности жидкостей и намного выше плотности газов (табл. 4.3).
Таблица 4.3 Плотность некоторых веществ, кг/м3

Исключением является вода, которая в твердом состоянии весит меньше, чем в жидком (табл. 4.4). А если бы она вела себя, как и все остальные вещества, то вода в морях и океанах промерзла бы насквозь, лед, будучи тяжелее воды, опустился бы на дно и лежал там не тая. И только на экваторе в небольшой толще воды существовала бы жизнь в виде нескольких видов бактерий.
Таблица 4.4. Плотность воды при разных температурах

Рефрактометрия — старейший оптический метод анализа, основы которого заложены Ньютоном, Эйлером и М. В. Ломоносовым. Метод базируется на измерении относительных показателей преломления (n).
Преломление света на границе двух сред — это изменение направления и скорости распространения светового луча. Если луч проходит из среды 1 (рис. 4.7) с меньшей преломляющей способностью в среду 2 с большей преломляющей способностью, то угол падения α будет больше угла преломления β.
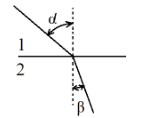
Рис. 4.7. Преломление света на границе двух сред
Относительный показатель преломления определяется по уравнению n2,1 = sinα / sinβ, где n 2,1 — относительный показатель преломления среды 2 по отношению к среде 1.
Показатель преломления можно выразить через скорости света (ν1, ν2) в средах 1 и 2:
n2,1 = ν1/ ν2. Показатель преломления луча, падающего на среду из безвоздушного пространства, называется его абсолютным показателем преломления или проcто показателем преломления данной среды (табл. 4.5).
Таблица 4.5. Показатель преломления некоторых сред

Аналитические возможности рефрактометрии:
Качественный анализ заключается в идентификации индивидуальных веществ. Показатель преломления — характерная для данного вещества константа. Рефрактометрически контролируют подлинность жидких лекарственных форм, эфирных масел, витаминов, сахарных сиропов.
Количественный анализ основан на зависимости показателя преломления от концентрации вещества. Рефрактометрически можно анализировать одно-, двух- и трехкомпонентные системы лекарственные препараты, спирты, сахара и др. Однако чаще всего проводят анализ двухкомпонентных растворов. Например, можно проводить количественный анализ солей в водных растворах (NaCl, NaBr, NaI, KBr, KI, CaCl2, MgSO4, NaHCO3, Na2S O3).
Достоинствами метода являются простота выполнения анализа и оборудования (рис. 4.8, 4.9); экспрессность и экономичность; для анализа требуется минимальное количество пробы.

К недостаткам метода можно отнести:
- ● низкую точность, которая существенно возрастает, если компоненты пробы будут иметь большую разницу в показателях преломления;
- ● низкую чувствительность (поэтому метод используется при анализе в области высоких концентраций (1 %);
- ● низкую селективность, обусловленную тем, что показатель преломления n — «неспецифическая» величина (для разных веществ значения n могут быть близки или даже совпадать).
Поэтому метод используется только для анализа индивидуальных веществ или простейших смесей, содержащих 2–3 компонента. Технические продукты всегда содержат примеси, которые влияют на величину показателя преломления. Поэтому он может в ряде случаев служить характеристикой чистоты продукта. Например, сорта очищенного скипидара различают по показателям преломления. Показатели преломления скипидара, обозначенные через n20D (запись означает, что показатель измерен при 20 °С, длина волны падающего света равна 598 мкм), равны:
1,469–1,472 — первый сорт;
1,472–1,476 — второй сорт;
1,476–1,480 — третий сорт.